
Page 133
4
Development and Regulation of New Technologies
DEVELOPMENT: THE PROCESS FOR SELECTING AREAS OF INVESTIGATION FOR CANCER DETECTION
Over the last decade, the activity and investment in research aimed at developing new technologies for early breast cancer detection have increased substantially, in part because of the efforts of advocacy groups and attention given by the U.S. Congress. At the same time, biomedical research has become more complex and capital intensive, requiring enormous investments to develop technologies and to generate and analyze data. Traditionally, basic research and the early stages of medical technology development were the realm of government-funded projects at universities or the National Institutes of Health, whereas private companies were primarily involved in bringing technologies to the market and the clinic. Although that may still be true in some instances, the lines between the various funders and developers of new technologies have blurred, and many new participants have also recently joined the process (see Table 1-1). As in many high-technology industries, the expanding development costs for new technologies and the growing importance of regulatory issues have provided powerful motives for public-private collaboration (National Research Council, 1999).
Many of the decisions about whether to pursue the development of novel and innovative technologies hinge on the perceived balance of opportunity and risk. Opportunity is determined by a combination of technological advances and the presence of an unmet need or interest in the market. Risks include considerations of the time and resources needed to
Page 134
develop technologies when the end results of the research and the profitability of the product are uncertain. In the case of medical technologies in particular, a large part of that uncertainty may be due to the additional processes required for Food and Drug Administration (FDA) approval (covered in this chapter) and adoption by the purchasers, providers, and users of medical care (covered in Chapters 5 and 6). A relative lack of patent protection can further diminish financial incentives and increase the risk for developing new technologies. New medical devices usually have some patent protection, but effective market exclusivity for devices is often shorter than the life of the patent because competitors can “invent around” device patents more easily than around new molecular entities like drugs (Medical Technology Leadership Forum, 1999). On the other hand, the relative lack of effective patent protection for devices provides incentives for other companies to invest in improving technologies already on the market. In fact, the original innovating company is rarely the sole or even the dominant source of further improvements.
NEW INITIATIVES AND COLLABORATIONS IN MEDICAL IMAGING RESEARCH
The limitations of mammography have been a driving force behind the search for new technologies that can detect breast cancer, eliminate unnecessary biopsies, and provide information that can be used to guide therapeutic decisions. Because of the multidisciplinary nature of technologies for breast cancer detection and diagnosis, a number of collaborative efforts have recently been established or explored (see Table 1-1).
In the spring of 1993, the National Cancer Institute (NCI) held a conference to explore the transfer of novel imaging technologies from the defense, intelligence, space, and energy communities for the purpose of breast cancer detection. The primary focus of that conference was technology transfer specifically for digital mammography and computer-aided detection (CAD). The following year, the U.S. Public Health Service's Office on Women's Health (OWH), along with the Central Intelligence Agency (CIA), NCI, FDA, and other federal agencies, established a working relationship with leaders of the medical imaging community to adapt defense technologies used for missile and target recognition to breast cancer detection. One example of such collaborations was between the University of Chicago and the National Information Display Laboratory, which led to advances in CAD technologies and played a major role in developing the current commercially available CAD software from R2 Technologies, Inc.
Productive interactions such as these led to further interest in applying new technologies to other imaging modalities, such as magnetic resonance imaging (MRI), ultrasound, and nuclear medicine. In the spring of
Page 135
1996, the Federal Multi-Agency Consortium to Improve Women's Health was established to formalize collaborations between the medical establishment and the nation's defense, intelligence, space, and energy communities. This new consortium included NCI, OWH, FDA, CIA, the National Science Foundation, the National Aeronautics and Space Administration, the U.S. Departments of Defense, Energy, and Commerce, and the Health Care Financing Administration. The initial goal of the consortium was to catalog the state of the art for breast imaging modalities and to identify the scientific and technological needs for application of the technologies to breast cancer detection, diagnosis, and treatment. These needs were then translated into a problem statement1 of technical specifications that could be understood by engineers, physicists, and other scientists working on imaging technologies. Since its inception, several national public workshops have been held to facilitate technology transfer and to stimulate public-private partnerships for technology development and application to breast cancer (Final Report, 1998).
Similar initiatives that focus more generally on biomedical imaging in oncology have also been launched recently. The first NCI-industry forum and workshop on this topic was held in September 19992 following discussions between NCI and the National Electrical Manufacturers' Association (NEMA).3 The forum had four main objectives: (1) to bring together individuals involved in funding, research, regulation, and reimbursement of imaging technologies; (2) to expand the role of anatomic and functional-molecular imaging in oncology; (3) to develop strategies for application of technical advances in imaging to unmet clinical needs in cancer; and (4) to better understand the processes related to development, adoption, approval, and dissemination of imaging technologies in oncology. A second workshop to continue the dialogue was held in September 2000.4
The first large-scale collaborative clinical trials group devoted to the
1http://www.4woman.gov/owh/bcimage/frames2.htm. Statements were prepared for digital X-ray mammography, MRI, ultrasound imaging, positron emission tomography, and nuclear medicine, computer-aided diagnosis, three-dimensional interactive visualization, and image storage and transmission. The problem statement for each technology describes the current state-of-the art as well as the technological needs and roadblocks to assist researchers unfamiliar with breast imaging in assessing the potential for their own technology, capability, or expertise to help address the stated needs.
2For a meeting summary, see
http://dino.nci.nih.gov/dctd/forum/summary.htm.
3NEMA is a trade organization for more than 600 companies, including 60 that produce diagnostic medical equipment.
4For a summary, see
http://dino.nci.nih.gov/dctd/forum/summary00.htm.
Page 136
development of technologies for medical imaging and the conduct of clinical trials to assess them was also launched in 1999. The American College of Radiology Imaging Network (ACRIN)5 has more than $22 million in funding from NCI for a 5-year period. ACRIN's fundamental objectives are to assess the value of emerging and established medical imaging tools by evaluating their effects on patient outcomes and costs, to increase participation in clinical trials, and to train researchers in conducting clinical trials.
The new multi-institutional consortium is structured to work with other NCI consortia, industry, and insurers. ACRIN has a more “virtual” organization compared with those for other multi-institutional study groups, with participating institutions at many distant sites contributing to an electronic database. The trial infrastructure integrates funding, methodological support, data acquisition and management, informatics, regulatory assistance, quality control, financial management, analysis, and research dissemination. All ACRIN clinical trials must be reviewed and approved by NCI's Cancer Therapy Evaluation Program.
ACRIN's recently approved screening mammography trial will compare full-field digital mammography with conventional mammography. The trial is about to begin, but investigators hope to accrue nearly 50,000 asymptomatic women at approximately 20 sites across the United States for screening mammography with digital machines from four manufacturers (Trex, Fischer, Fuji, and General Electric). The study participants will not be randomized, and all women will undergo both conventional and digital mammography. Two readers will examine the images independently, and decisions for diagnostic workup will be based on the results of both examinations. As designed, the study should have adequate statistical power to determine whether unnecessary recalls for follow-up tests can be reduced by use of digital mammography. The major outcomes measures will include the sensitivity, specificity, positive predictive value, and negative predictive value of digital mammography. Secondary goals are to compare the accuracy of mammography using “soft-copy” image display to that of laser-printed films and to examine the effect of breast density on accuracy. The study is expected to be completed by 2004.
Several initiatives and collaborations aimed at characterizing the molecular biology of cancer and identifying tumor markers have also been launched recently, as described in more detail in
Chapter 3. A prime example is the Cancer Genome Anatomy Project of NCI, whose goal is to develop a comprehensive molecular characterization of normal, precancerous, and malignant cells. NCI also recently announced a novel pro-
5www.acrin.org.
Page 137
gram that will study the stages of breast development, from normal development of the mammary gland to the metastatic changes of breast cancer.6 This program encourages multidisciplinary collaborations among such specialists as cell and molecular biologists, bioengineers, geneticists, and pathologists.
EXAMPLES OF FUNDING MECHANISMS FOR MEDICAL TECHNOLOGY DEVELOPMENT
National Cancer Institute
Historically, NCI has conducted and funded basic, applied, and clinical and health services research to acquire new knowledge that can be used to prevent, diagnose, and treat cancer (Tables 4-1 and 4-2). Recently, NCI has also developed programs to make its peer review system, which has traditionally been based on hypothesis-driven science, more accessible for technology development. Two years ago, NCI established the Office of Technology and Industrial Relations, with the goal of facilitating expedited technology development and transfer activities. The office oversees the Small Business Innovative Research (SBIR) awards and the Small Business Technology Transfer Research (STTR) grants. The SBIR and STTR programs are designed to support innovative research that has the potential for commercialization. In the case of SBIR awards, the research is conducted solely by small businesses, whereas STTR grants support research conducted cooperatively by a small business concern and a research institution. For the latter, the small business must carry out at least 40 percent of the research project and the partner research institution must perform at least 30 percent of the work.
Support under the SBIR-STTR programs normally includes $100,000 for 6 months for Phase I (proof of concept) and $750,000 for 2 years for Phase II (development). However, the Phased Innovation Award is a new mechanism directed at supporting research on new technologies, from the evolution of the innovative concept to the research development phase. Compared with the traditional two-step grant application process, which can be cumbersome, the Phased Innovation Award allows a single grant application for two previously distinct awards. The new award permits flexible research programs for up to 4 years. To move into the development phase, investigators must achieve measurable milestones.
Areas of focus include cancer imaging and definition of the molecular changes, or signatures, of tumors. A particular focus has been placed on technologies that will permit multiple levels of analysis: in vitro (test
6http://grants.nih.gov/grants/guide/pa-files/PA-99-162.html.
Page 138
|
Breast Cancer Researcha |
Breast Cancer Detectionb |
|||||
|
Fiscal Year |
Grants |
Contracts |
Total Extramural |
Grants |
Contracts |
Total Extramural |
|
1999 |
325,496,000 |
11,477,000 |
336,973,000 |
58,520,698 |
1,571,010 |
60,091,708 |
|
1998 |
289,621,000 |
10,738,000 |
300,359,000 |
52,642,462 |
636,025 |
53,278,487 |
|
1997 |
269,742,000 |
17,721,000 |
287,463,000 |
51,297,401 |
1,999,580 |
53,296,981 |
|
1996 |
226,720,000 |
5,212,000 |
231,932,000 |
45,967,762 |
4,192,250 |
50,160,012 |
|
1995 |
183,684,000 |
10,293,000 |
193,977,000 |
46,638,799 |
1,583,246 |
48,222,045 |
|
1994 |
171,706,000 |
20,798,000 |
192,504,000 |
40,312,325 |
757,661 |
41,069,986 |
|
1993 |
143,671,000 |
17,836,000 |
161,507,000 |
33,189,587 |
1,704,912 |
34,894,499 |
|
1992 |
19,872,037 |
2,687,428 |
22,559,465 |
|||
|
1991 |
17,063,894 |
1,119,392 |
18,183,286 |
|||
|
1990 |
14,486,403 |
1,928,983 |
16,415,386 |
|||
a
Includes the three categories indicated, as well as training and basic research and other scientific areas.
b
Any project involving actual detection of breast tumors (screening, diagnosis, clinical trials), development or refinement of diagnostic techniques or devices, education, or promotion of breast cancer detection.
c
Anything designed to inhibit the onset of breast cancer, such as drug administration or lifestyle changes.
tube), in situ (cellular), and in vivo (the whole body). NCI is also developing programs that aim to bring the field of molecular biology together with the imaging community, with the goal of identifying the fundamental molecular changes in tumors. Several program announcements address this area, including developmental grants for diagnostic cancer imaging and the study of novel imaging modalities. NCI has also promoted the study of new imaging agents and probes, especially those that have the potential to better pinpoint the molecular signatures of tumors.
To further spur development of innovative and high-risk technological improvements in cancer detection and treatment, the Office of Technology and Industrial Relations has also developed a nontraditional,
Page 139
|
Breast Cancer Preventionc |
Breast Cancer Treatment |
||||
|
Grants |
Contracts |
Total Extramural |
Grants |
Contracts |
Total Extramural |
|
27,867,000 |
2,461,799 |
30,328,799 |
85,705,732 |
1,965,658 |
87,671,390 |
|
27,363,273 |
4,701,252 |
32,064,525 |
67,498,736 |
982,960 |
68,481,696 |
|
30,413,527 |
143,282 |
30,556,809 |
77,072,791 |
336,089 |
77,408,880 |
|
20,366,677 |
2,164,405 |
22,531,082 |
77,504,241 |
1,214,610 |
78,718,851 |
|
18,883,694 |
1,556,852 |
20,440,546 |
69,572,849 |
1,887,207 |
71,460,056 |
|
16,039,590 |
3,281,600 |
19,321,190 |
58,987,022 |
4,219,795 |
63,206,817 |
|
20,175,725 |
2,318,649 |
22,494,374 |
53,190,603 |
5,801,301 |
58,991,904 |
|
18,891,894 |
3,822,326 |
22,714,220 |
40,935,757 |
653,008 |
41,588,765 |
|
10,739,031 |
2,582,304 |
13,321,335 |
24,426,247 |
724,750 |
25,150,997 |
|
9,756,434 |
1,398,349 |
11,154,783 |
24,874,428 |
1,853,666 |
26,728,094 |
SOURCES: NCI. Anna Levy, NCI Office of Women's Health; Marilyn Gaston, NCI Inquiry and Reporting Section; Rosemary Cuddy, NCI Division of Extramural Activities.
multidisciplinary program through a new program, the Unconventional Innovations Program.7 In addition to recruiting new investigators and building multidisciplinary teams, the program is actively engaged in translating technology into other nontraditional domains. The program is modeled after the Defense Advanced Research Projects Agency and emphasizes technology maturation and dissemination.
This program solicits contracts for the development of novel technologies for noninvasive detection, diagnosis, and treatment of cancer.
7http://www.nih.gov/news/pr/oct98/nci-06a.htm.
Page 140
|
Imaging |
Nonimaging |
|||||
|
Fiscal Year |
Grants |
Contracts |
Total |
Grants |
Contracts |
Total |
|
1999 |
38,099,604 |
916,001 |
39,015,605 |
20,421,094 |
655,009 |
21,076,103 |
|
1998 |
37,587,304 |
93,716 |
37,681,020 |
15,055,158 |
542,309 |
15,597,467 |
|
1997 |
38,884,336 |
1,999,580 |
40,883,916 |
12,413,065 |
0 |
12,413,065 |
|
1996 |
37,993,657 |
2,363,847 |
40,357,504 |
7,974,105 |
1,828,403 |
9,802,508 |
|
1995 |
37,179,948 |
1,566,752 |
38,746,700 |
9,458,851 |
16,494 |
9,475,345 |
|
1994 |
31,135,248 |
590,009 |
31,725,257 |
9,177,077 |
167,652 |
9,344,729 |
|
1993 |
24,500,571 |
1,604,231 |
26,104,802 |
8,689,016 |
100,681 |
8,789,697 |
|
1992 |
16,770,171 |
1,974,525 |
18,744,696 |
3,101,866 |
712,903 |
3,814,769 |
|
1991 |
13,138,486 |
933,272 |
14,071,758 |
3,935,408 |
186,120 |
4,121,528 |
SOURCES: NCI. Anna Levy, NCI Office of Women's Health; Marilyn Gaston, NCI Inquiry and Reporting Section; Rosemary Cuddy, NCI Division of Extramural Activities.
Page 141
According to the most recent solicitation announcement,8 the program is “specifically soliciting projects to develop technology systems or systems components that will enable the sensing of defined signatures of different cancerous and precancerous cell types or their associated microenvironment in the body in a way that is highly sensitive and specific, yet noninvasive.” The highest priority is for systems that can “either support or provide a seamless interface between sensing/detection and intervention.” The stated goal of the program is to “develop technology that will target quantum improvements in existing technologies or entirely novel approaches, rather than incremental improvements to state of the art.” The first five awards totaling $11.3 million were issued in 1999 (Table 4-3). Within the next 5 years NCI plans to invest $48 million through this program.
Because of the tremendous need for information systems development in the area of high-throughput biological analysis, NCI is also actively building the Biomedical Information Science and Technology Implementation Consortium (BISTIC)9 to promote database creation and management that will permit modeling, manipulation, and hypothesis generation. Key components of the strategy include supporting planning grants in national centers of excellence, furthering investigator-initiated research, bolstering the Information Storage, Curation, Analysis, and Retrieval program, coordinating work across the National Institutes of Health (NIH), and building a computing infrastructure. Initially, investments will focus on traditional, low-risk, evolutionary projects, but BISTIC will move toward long-term, higher-risk investments. NCI is also supporting the development of modeling tools to facilitate the identification of new targets for detection and intervention.
In the spring of 2000, NIH combined BISTIC with the Bioengineering Consortium to create the Office of Bioengineering, Bioimaging, and Bioinformatics (OBBB) (Haley, 2000). Recently, legislation required NIH to establish a new institute, the Institute for Biomedical Imaging and Bioengineering.10 This legislative action has effectively terminated OBBB, which would have operated in conjunction with the current disease-oriented institutions by providing a coordinating infrastructure for bio-imaging and bioengineering initiatives. OBBB did not have grant-making authority or funds itself, in contrast to the new institute, which will have such authority (Softcheck, 2001). However, the field of bioinformatics was not included in the scope and mission of the new institute.
8http://Amb.nci.nih.gov/UIP.HTM.
9http://grants.nih.gov/grants/bistic/bistic2_t.htm.
10A bill to create the new NIH Institute for Biomedical Imaging and Bioengineering, H.R. 1795, was signed into law on December 29, 2000, by President Bill Clinton (Haley, 2000).
Page 142
|
Summary of 1999 Projects for Noninvasive Detection, Diagnosis, and Treatment of Cancer |
Amount Awarded |
|
|
1. |
The project will focus on the development of two specific devices, based on current polymer technology, for detection and treatment of cancer; one will be useful for detection of adenocarcinoma of the breast, whereas the other will be used for detection of adenocarcinoma of the colon. |
$4.4 million |
|
2. |
The project will develop a novel optical technique using near infrared for identification of precancer and early cancers. Among other things, it is expected that the end product of the technology will yield a noninvasive (nonintrusive) method of detection for small human breast cancers. |
$2 million |
|
3. |
The project targets the development of a novel system capable of selective transduction (a potential avenue of gene therapy) of target cells in the context of the clinical settings for posttreatment recurrent neoplastic disease and metastatic disease and early subclinical or undetected preneoplastic disease. |
$1.8 million |
|
4. |
The aim of the project is to construct and demonstrate a prototype compact device that may be used in a variety of biomedical applications including in vitro and in vivo spectroscopy and high-resolution imaging, mammography, diagnosis, and radiation therapy. |
$1.6 million |
|
5. |
This project will develop novel carbon nanotube-based biosensor technology and develop a prototype biosensor catheter, suitable for in vitro use, that permits detection of specific oligonucleotide sequences that serve as molecular signatures of cancer cells. |
$1.5 million |
SOURCES: http://otir.nci.nih.gov/tech/vip_awards.html, and Richard Hartman, contracting director, NCI, personal communication (July, 2000).
U.S. Department of Defense
The U.S. Department of Defense (DOD) is the second largest administrator of federally funded breast cancer research in the United States. In addition to managing biomedical research programs that are part of the Army and DOD budget submission, the U.S. Army Medical Research and Materiel Command (USAMRMC) can be directed by the U.S. Congress to undertake special-interest biomedical research programs. One of the congressional research programs managed by USAMRMC is the Breast Cancer Research Program (BCRP),
established11
in 1992 with $25 million ap-
11BCRP was established by Joint Appropriations Bill 102-328.
Page 143
propriated for research on breast cancer screening and diagnosis for military personnel and their dependents. In response to requests from breast cancer advocacy groups (Marshall, 1993), the 1993 Defense Appropriations Act provided $210 million for a “peer-reviewed breast cancer research program with the Department of the Army as the executive agent.” Appropriations for fiscal years 1992 through 2000 total $1.043 billion. In 1995, $20 million (of the total $150 million appropriation) was earmarked for research in mammography and breast imaging with the goal of applying military technology to mammography to improve its accuracy.
The objective of BCRP is to fund the training of new scientists in the field, infrastructure enhancement, and investigator-initiated research with a balanced portfolio of research on the prevention, detection, diagnosis, and treatment of breast cancer ( Table 4-4). Since its inception, the program has funded research at U.S. and international universities, hospitals, nonprofit and for-profit institutions, private industry, and state and federal agencies. BCRP has developed a funding strategy (Institute of Medicine, 1993, 1997) with the goal of complementing awards made by other agencies and has specifically tried to avoid duplication of long-term basic research supported by NIH. Research awards have been made by a twotiered review process that includes consumer advocates. Peer review panels assess the scientific merit of research proposals, whereas the programmatic reviews determine the contribution of projects to the program goals. BCRP was the first program to use this format for review of grant applications, and since its adoption, NCI has also become more open to the participation of consumer advocates.
BCRP has also initiated new approaches to grant applications through two new awards: the Idea Award and the Concept Award. The intent of the Concept Award is to provide initial funding for a novel concept or theory that could give rise to a testable hypothesis. The one-page Concept Award proposals go through a fast-track submission and review process. Preliminary data are not required, and the award provides $50,000 for 1 year. The Idea Award also does not require preliminary data and is designed to foster innovative ideas and technologies that ‘challenge the existing paradigms'. The average grant in this category provides $100,000 per year for up to 3 years.
Advanced Technology Program, National Institute of Standards and Technology
The National Institute of Standards and Technology (NIST) is an agency of the U.S. Department of Commerce's Technology Administration. NIST works with industry to develop and apply technology, mea-
Page 144
|
Prevention |
Early Detection, Prognosis |
||||
|
Year |
Congressional Appropriationa |
No. of Awards |
Dollars |
No. of Awards |
|
|
1992-1994 |
$235.0 |
10 |
$2,654,443 |
97 |
|
|
1995 |
$150.0 |
8 |
$2,711,964 |
77 |
|
|
1996 |
$75.0 |
7 |
$1,519,845 |
58 |
|
|
1997 |
$108.3 |
9 |
$2,678,355 |
69 |
|
|
1998 |
$135.0 |
21 |
$7,075,114 |
106 |
|
|
Total |
$703.3 |
55 |
$16,639,721 |
407 |
|
NOTE: If both the first and second choices of category for a grant were in the same main category, the funding was counted only once; if the codes were in two different categories, the full amount was counted in both categories.
aIn millions of dollars.
surements, and standards. It carries out this mission through four major programs, including the Advanced Technology Program (ATP).12
The goal of ATP is to stimulate innovation by bridging the gap between the research laboratory and the marketplace. Through partnerships with the private sector, ATP's early stage investments aim to accelerate the development of novel technologies that promise significant and widespread benefits for the nation, in addition to a direct return to the innovators. By sharing the relatively high risks of developing new technologies, ATP can foster projects with greater technical challenges that companies could not or would not take on by themselves.
The ATP has several critical features that set it apart from other government research and development programs:
-
Projects focus on the technology needs of U.S. industry, not those of the U.S. government.
-
Research priorities are set by industry on the basis of their understanding of the marketplace and research opportunities.
-
For-profit companies conceive, propose, cofund, and execute projects and programs in partnerships with academia, independent research organizations, and federal laboratories.
-
Strict cost-sharing rules are in place.13
12http://www.atp.nist.gov/atp/overview.htm.
13Joint ventures (two or more companies working together) must pay at least half of the total project costs. Large, Fortune 500 companies participating as a single firm must pay at least 60 percent of the total project costs. Small and medium-sized companies working on single-firm ATP projects must pay a minimum of all indirect costs associated with the project.
Page 145
|
Diagnosis, and |
Treatment |
||
|
Dollars |
No. of Awards |
Dollars |
Total No. of Awards |
|
$52,666,720 |
50 |
$26,520,447 |
157 |
|
$25,981,647 |
44 |
$19,900,740 |
129 |
|
$11,034,618 |
61 |
$13,759,763 |
126 |
|
$22,305,064 |
79 |
$29,906,133 |
157 |
|
$22,962,976 |
88 |
$25,774,586 |
215 |
|
$134,951,025 |
322 |
$115,861,669 |
784 |
SOURCE: Office of Congressionally Directed Medical Research Programs, Medical Research and Material Command (Stacey Young-McCaughan, Deputy Director).
-
ATP does not fund product development. Private industry bears the costs of product development, production, marketing, sales, and distribution.
-
Awards are made strictly on the basis of rigorous peer-reviewed competitions. Selection is based on the innovation, the technical risk, the potential economic benefits to the nation, and the strength of the commercialization plan for the project.
-
Support does not become a perpetual subsidy or entitlement. For each project goals, specific funding allocations, and completion dates are established at the outset. Projects are monitored and can be terminated for cause before completion.
ATP has funded companies of all sizes, many of which were involved in partnerships with universities and nonprofit organizations. To date, more than half of the ATP awards have gone to individual small businesses or to joint ventures led by a small business. Well over half of the ATP projects to date also include one or more universities as either sub-contractors or members of a joint venture. ATP awards are selected through open, peer-reviewed competitions. All industries and all fields of science and technology are eligible. Proposals are evaluated by technology-specific boards that are staffed with experts in fields such as
Page 146
biotechnology, photonics, chemistry, manufacturing, information technology, and materials.
ATP has funded several projects that may have a potential effect on breast cancer. Some of these have focused on increasing the efficiency and imaging capabilities of technologies such as X ray, MRI, ultrasound, and other imaging devices while lowering their costs. As one example, ATP awarded $6 million over 4 years to Xerox, in conjunction with Thermotrex Corp., for a proposal to improve the resolution and accuracy of a digital imaging system. ATP has also heavily funded genomic and biotechnology research projects that may have an effect on breast cancer screening, diagnosis, and treatment. For example, ATP awarded Affymetrix and Molecular Dynamics $31 million to develop a ‘miniature nucleic acid diagnostic device', which consists of DNA arrays on silicon chips, with the goal of providing a rapid and accurate means of diagnosis of a wide variety of diseases, including breast cancer.
PRIVATE INVESTMENT
The large, established medical imaging companies devote significant resources to research and development aimed at improving imaging technologies that are already in use and that have broad applications, such as MRI, ultrasound, and X-ray modalities. However, small start-up companies often initially pursue novel technologies, especially those that involve higher risk and potential benefits. About 80 percent of medical device companies are start-ups with less than 50 employees and thus have limited resources at their disposal to generate data and evidence for approval. The devices may cost thousands of dollars each, making distribution of devices for the purpose of conducting clinical trials very expensive. As a result, the timing of some trials may be delayed and the protocols may be restricted. Data collection may also be curtailed by limited patient follow-up or the number of end points assessed (Medical Technology Leadership Forum, 1999).
Investment of venture capital is one way of providing nongovernment start-up monies for the development of new medical technologies. Typically, venture capital firms raise capital, make investments in a small start-up company, and then sell the assets when the product goes into the market. Venture capitalists generally look for a proprietary product with a large market, clearly identified customers, and minimal impediments to adoption and diffusion. In other words, when the perception of risk is low and the returns are expected to be high, investment is more probable.
Traditionally, a venture capitalist's investment timeline was 4 to 10 years. The investment portion of the cycle took about 5 years, with another 3 or 4 years needed to harvest the assets. The expectations for that cycle have changed radically in the last 2 years, largely because of the
Page 147
Internet, which has spurred enormous investments and exceptional rates of return. On average, it takes an Internet-related business less than 2 years to go public, whereas previously the norm was 3 to 5 years. Because initial public offerings happen much more quickly with Internet-related businesses, investors' money is not locked up as long as it is in traditional investments. In addition, venture capital investors have been earning returns in excess of 100 percent on their Internet-related investments (PricewaterhouseCoopers, 2000). Although these recent trends may not continue in the future, the global economic environment is driving technology research in the private sector. The pace of technological change in general is faster than ever before, compelling companies to make narrower, shorter-term investments in research and development that maximize returns quickly.
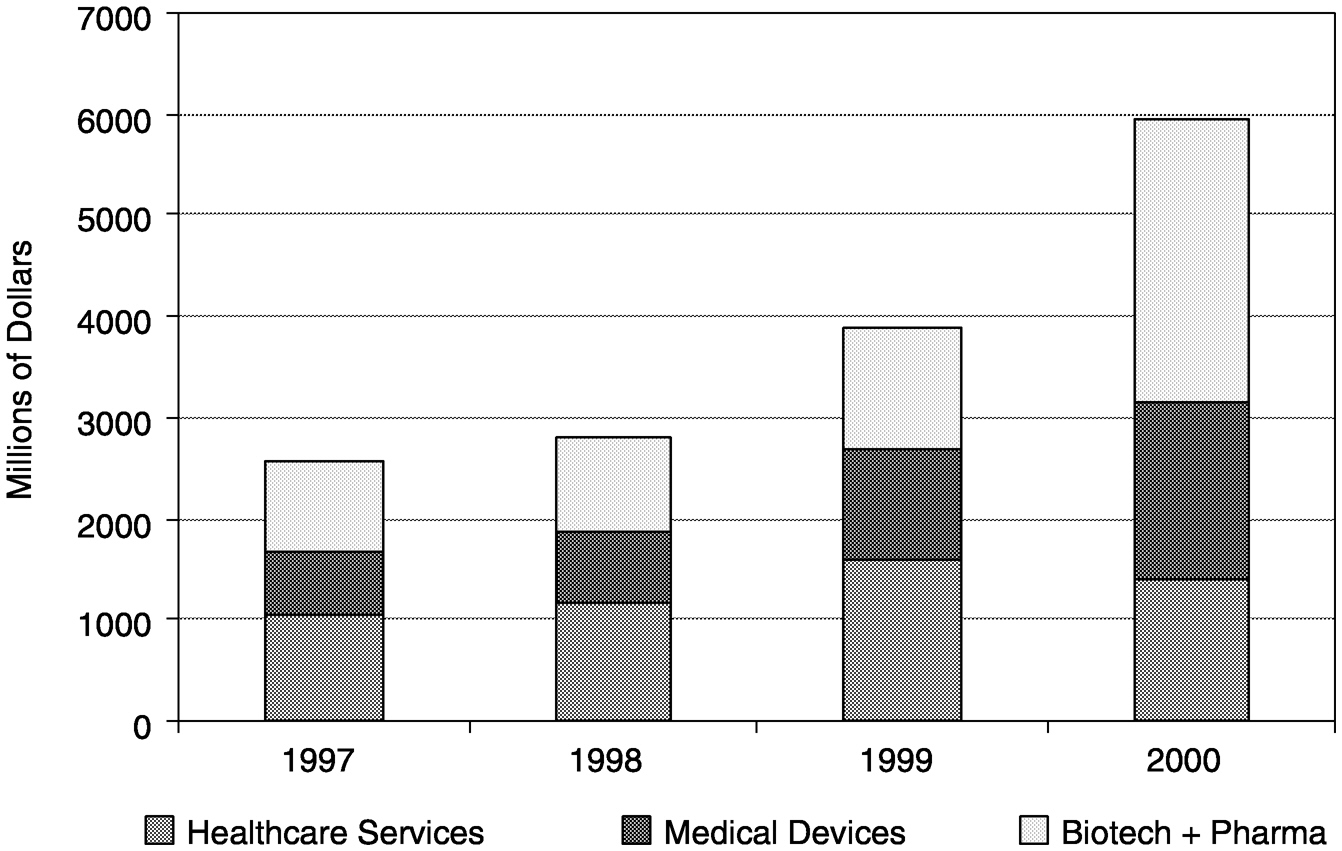
These recent investment trends have not been favorable to the health care industry, which typically has a longer investment cycle and more modest returns. Although actual venture capital investments in health care-related companies were at an all-time high in 1999 (Figure 4-1), this increase was largely driven by health care-related Internet endeavors. During the last quarter of 1999, 68 percent of the venture capital cash invested in health care-related companies was devoted to such “e-health” stocks. In 2000, more than 50 percent of venture capital money in health
~ enlarge ~
FIGURE 4-1 Venture capital investments in health care industries, 1997 to 2000. SOURCE: PricewaterhouseCoopers Money Tree™ Survey.
Page 148

~ enlarge ~
FIGURE 4-2 Health-care related industry investments (includes health-care services, biotechnology, pharmaceuticals, and medical devices) as a percentage of total venture capital investments, 1997 to 2000. Source: PricewaterhouseCoopers Money Tree™ Survey.
care was still going to internet-related ventures. Furthermore, the percentage of total venture capital investment going into health care has plunged from about 25 percent traditionally to about 8 percent by the end of 2000 (Figure 4-2). Only 2.5 percent of all 2000 venture capital investments was devoted to medical device companies (Figure 4-3).14
Venture capital firms may view investment in the diagnostic and device areas of the health care sector as a high-risk proposition because of the length of time required to produce data for FDA approval and insurance coverage decisions and because of the uncertainty associated with those decisions. Unfavorable reimbursement levels and slow adoption rates for new technologies can add further complications. Technologies that improve on existing screening and diagnostic modalities may be viewed as particularly risky with regard to the latter because competition in the marketplace may lead to inadequate returns on investment.
In 1999, estimated U.S. expenditures on diagnostic imaging and therapy systems were approximately $5 billion dollars, whereas those for
14Data were obtained from PricewaterhouseCoopers Money Tree™ Survey for 1999. The survey measures venture capital equity investments in the United States on a regional and a national basis and serves as a barometer of economic health through entrepreneurial developments.
Page 149
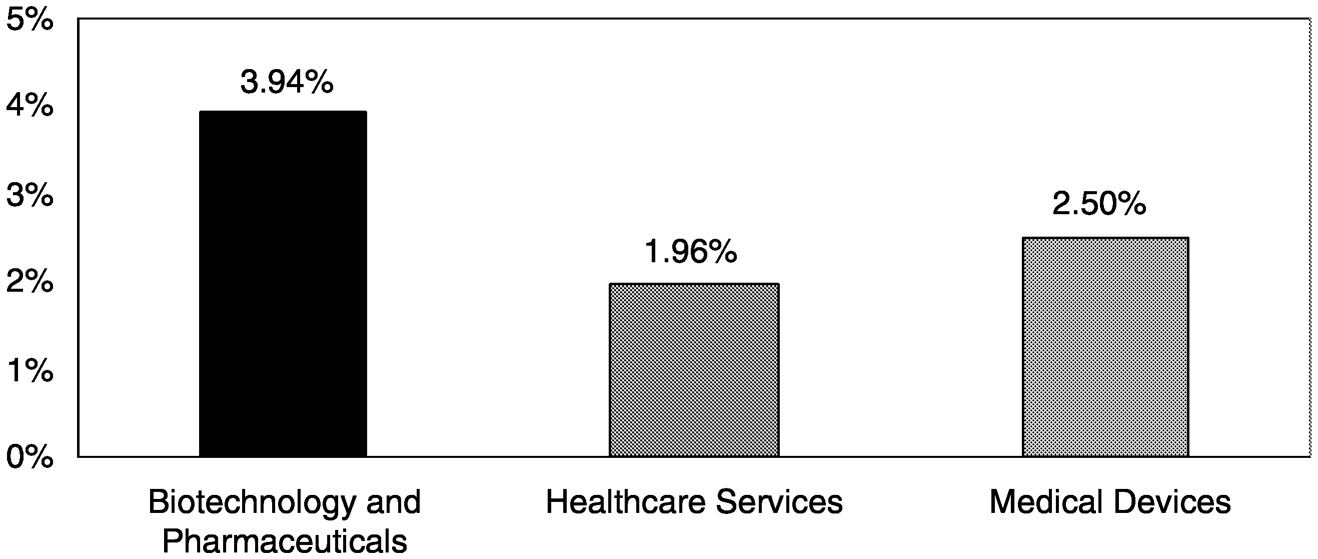
~ enlarge ~
FIGURE 4-3 Percentage of total venture capital investments in specific healthcare industries, 2000. SOURCE: PricewaterhouseCoopers Money Tree™ Survey.
drugs were $120 billion15 (Table 4-5). Ultrasound and X-ray equipment accounted for about 20 and 30 percent of the total, respectively. However, within each of those categories only a small fraction entails breast imaging equipment (the United States has approximately 10,000 certified breast imaging centers16). Because it can cost technology sponsors many millions of dollars to bring a new technology to market, the opportunity for a sizeable return on investment is thus relatively small.
FDA APPROVAL: PROCESS FOR GETTING NEW TECHNOLOGIES INTOTHE CLINIC
FDA is charged with regulating the “safety and effectiveness” of all medical products sold within the United States. Hence, FDA has jurisdiction over all drugs, devices, and biologies, but its approach varies for each category. To understand the specific issues of FDA approval of technologies for breast cancer screening and diagnosis, it is necessary to first look at the general processes used by FDA. Technologies for the early detection of breast cancer will most likely involve either imaging devices or clinical laboratory devices, so the general description of FDA procedures provided here will focus briefly on these categories, which are overseen by the Center for Devices and Radiological Health of FDA.17
15Total retail prescription drug sales in 1999 were $121.7 billion
(http://www.nacds.org/; accessed December 15, 2000).
16See
http://www.fda.gov/cdrh/mammography/mqsa_accomplisnments.html
17www.FDA.gov/cdrh/. Some imaging techniques, such as scintimammography, may require review by FDA's Center for Drug Evaluation and Research because they make use of radionuclide agents.
Page 150
|
Sales Billed |
1991 |
1992 |
1993 |
1994 |
1995 |
1996 |
1997 |
1998a |
1999a |
|
Computed tomography |
453 |
477 |
453 |
372 |
396 |
525 |
543 |
594 |
663 |
|
MRI |
717 |
724 |
665 |
691 |
625 |
620 |
592 |
729 |
842 |
|
Nuclear imaging |
102 |
194 |
240 |
313 |
337 |
307 |
363 |
360 |
331 |
|
Radiation therapy |
488 |
461 |
431 |
388 |
436 |
540 |
576 |
535 |
551 |
|
Ultrasound |
708 |
877 |
788 |
784 |
782 |
947 |
1,086 |
1,126 |
1,083 |
|
X ray |
1,615 |
1,144 |
1,710 |
1,755 |
1,850 |
1,529 |
1,655 |
1,767 |
1,611 |
|
Total |
4,083 |
3,877 |
4,288 |
4,303 |
4,425 |
4,467 |
4,816 |
5,112 |
5,081 |
a Based on estimates and projections provided by the NEMA Economics Department.
SOURCES: Current Industrial Reports, U.S. Bureau of the Census, U.S. Department of Commerce.
Page 151
The Medical Device Amendments of 1976 established a device classification system based on the risk associated with each device and the ability to control that risk.18 The intent of the amendments was to provide “reasonable assurance” of the safety and effectiveness of medical devices. There are now three classes of devices with each class having increasing levels of FDA review and control (Box 4-1). All medical devices legally marketed before adoption of the amendments were classified into one of these three classes (Class I, II, or III). With the exception of some low-risk devices (most Class I devices, as well as a few Class II devices, are exempt from premarket review), medical devices must now undergo an FDA review before being introduced into the market. There are two major pathways to FDA approval, known as “premarket notification” [510(k)] or “premarket approval” (PMA) application (Box 4-2). Most devices are cleared for commercial distribution in the United States by the premarket notification process [510(k)].
Before undertaking clinical trials for FDA approval, a company must submit a plan explaining how it will conduct the clinical study, including a statement of objectives for the trial, what results are expected, and what risks and precautions may be involved. If FDA19 considers the request to be sound, it will grant an investigational device exemption (IDE), which allows the device to be used on patients, with informed consent, only for the purpose of gathering data in a clinical trial.20 In recent years, FDA has also encouraged sponsors to undergo a study protocol review process in which FDA engages in an interactive critique of the experimental design with respect to how a particular study fits into the sponsor's plans for development of the data for marketing approval or clearance (Sapirstein, 2000).
18Before that, FDA regulated devices under the adulteration and misbranding provisions of the Federal Food, Drug, and Cosmetic Act of 1938 (21 U.S.C.§321 et seq.), but premarket clearance was not required. When FDA learned that a device was unsafe or ineffective, it had to undertake time-consuming and costly legal action to prove that the device was unsafe or that the manufacturer's claims were not true. During the 1960s many new and complex devices entered the market, and FDA found it increasingly difficult to protect patients using this approach (Kahan, 1995). As a result, the Medical Device Amendments Act of 1976 required FDA to set standards for some devices and to undertake premarket clearance for others, although devices with minimal risks are exempt from both requirements.
19Institutional review boards (IRBs) may also play a role in determining whether an IDE is granted. A study approved under IDE, with informed consent, still requires IRB oversight.
20In the case of in vitro diagnostic studies, it is possible in some instances to perform the study without an IDE, especially if test results are blinded and not used clinically. Informed consent and IRB oversight may still be required, however, particularly if clinical information is linked to patient results.
Page 152
BOX 4-1
|
The evidence gathered for FDA approval must reflect the intended use and labeling of the device. For instance, manufacturers must state whether the device is intended for screening or diagnosis or for a specific population rather than a general population. Approval by FDA may be accompanied by a requirement for additional postmarket analysis to assess performance standards. For example, a company may be asked to measure the sensitivity and specificity of a device in clinical practice for specific populations or under specific conditions.
In 1997, the Food and Drug Administration Modernization Act (FDAMA) responded to industry concerns that the FDA process was burdensome. Under FDAMA, there is generally more flexibility and a more interactive exchange between FDA and the sponsor, allowing more rapid communications with companies.
EVALUATION OF SCREENING AND DIAGNOSTIC DEVICES
In many ways, film-screen mammography (FSM) does not provide a useful model of the development and approval process for screening and
Page 153
BOX 4-2
|
diagnostic technologies that are under investigation. The first major clinical trial to test the effect of mammography screening on clinical outcome was organized and supported by a health insurance provider (the Health Insurance Plan of Greater New York), not a device manufacturer (Shapiro, 1997). Furthermore, because X-ray mammography was already in clinical use at the time of passage of the Medical Device Amendments of 1976, it was not required to undergo a rigorous FDA approval process to demonstrate safety and effectiveness (rather, it was “grandfathered [for more detail, see below and Chapter 5]). In fact, since its inception, FDA's Center for Devices and Radiological Health has reviewed relatively few cancer screening technologies. Thus, there is a lack of precedent for defining the appropriate experimental strategies for FDA approval of such screening technologies.
Page 154
In some ways, the approval process for screening and diagnostic devices and tests can be more complicated than that for therapeutics because each device goes through a unique development process and the requirements of FDA can vary considerably for different devices. The stages of development for drugs, in contrast, are more standardized (Table 4-6). A similar paradigm for diagnostic devices has been developed (Table 4-7),21 but the specific end points of each phase are not well defined.
Therapeutic evaluation is more straightforward because the study end points are well defined. In general, therapeutic interventions generate direct outcomes that can be observed in individuals. In contrast, most patient-level effects of diagnostic devices are mediated by subsequent follow-up or therapeutic decisions. Diagnostic tests generate information, which is only one of the inputs into the decision-making process. Hence, the evaluation of diagnostic tests is fundamentally the assessment of the value of information. “Valuable” information should be obtainable easily and reliably, should be accurate, and should have the potential to influence the process of health care toward better outcomes.
Ideally, screening and diagnostic modalities would be deemed effective if they led to a reduction in disease-specific mortality rates (or perhaps reduced levels of morbidity and an enhanced quality of life). However, FDA approval of detection technologies generally does not focus on clinical outcomes (Houn et al., 2000). The clinical trials necessary to measure such a reduction in mortality must include a large number of subjects, be very long in duration, and thus are very expensive and arduous to undertake. The alternative is to measure the sensitivity and specificity of the modality, assuming that the efficacy and effectiveness of a device are determined by these two values (which may or may not be true).
To some extent, “effectiveness” can depend on the context in which the device will be used. There is often a trade-off between a device's sensitivity and specificity, and choosing which one to optimize may depend on whether it is used for screening or diagnosis, or both. In evaluating a new screening device, it may be more important to have a high sensitivity (the test's ability to detect everyone with cancer), whereas a diagnostic device may require a higher specificity (so that cancer is unlikely in those who do not test positive). If different methods are used for screening and diagnosis, then the high sensitivity of the screening step could be complemented by the high specificity of the diagnostic step. However, if the specificity of the screening device is too low, there will be problems associated with the cost and anxiety generated as a result of too many unnecessary follow-up diagnostic tests.
Safety can also have multiple definitions. For most devices, safety is
21Constantine Gatsonis, Brown University, manuscript in preparation.
Page 155
|
Trial Phase |
Design, Objective, and Endpoint |
|
Preclinical studies |
General safety and effect of the drug is tested in animals and in vitro. |
|
Clinical Phase I |
Small studies (∼10–100 participants) designed to determine the maximum safe dose of drug for use in humans. Drug doses start out very low and escalate as the study progresses. Trial participants are monitored for adverse side effects and drug level. |
|
Clinical Phase II |
Moderately sized (∼50–500 participants) studies designed to establish preliminary estimates of effective drug doses and duration. A primary goal is to determine the appropriate protocol (experimental conditions and end points) for the final phase of testing. The effects of the drug in study subjects are measured in comparison with those in control subjects. |
|
Clinical Phase III |
Very large studies (several hundred to several thousand participants) designed to definitively determine the effects of drug treatment. Investigators measure drug efficacy and side effects in study subjects compared with those in a control group of subjects. |
|
Clinical Phase IV |
Very large studies (several thousand participants) designed to assess effectiveness in clinical practice, assess cost-effectiveness, or test new indications for a drug that is already on the market. |
|
Trial Phase |
Design, Objective, and Endpoint |
|
Stage I (discovery) |
Establishment of technical parameters and diagnostic criteria |
|
Stage II (introductory) |
Early quantification of diagnostic accuracy |
|
Stage III (mature) |
Comparative assessment of accuracy and outcome in large clinical studies (efficacy). |
|
Stage IV (disseminated) |
Assessment of the procedure as utilized by the community at large (effectiveness). |
SOURCE: Constantine Gatsonis, Brown University, personal communication, August, 2000
Page 156
determined by whether use of the device itself can directly cause harm to the patients (for example, by exposing the patient to ionizing radiation). However, in the case of screening and diagnostic devices, harm can also result from missed detection and diagnosis (false-negative results), unnecessary follow-up procedures (false-positive results), or unnecessary treatment (overdiagnosis) as a result of using a device. Does this also fall under the purview of FDA regulations? The answer seems to vary, depending on the device in question, as will be discussed further in the case studies below. For clinical laboratory tests, FDA evaluation specifically does not examine these issues (i.e., an assessment of how the information generated by the test will be used) but, rather, entails only an assessment of the test's accuracy and reliability in detecting the analyte of interest.
In the case of diagnostic and screening modalities for breast cancer, the traditional notion of equivalence between technologies is also difficult to define solely in terms of the average performance of the devices. The clinical evaluation of these technologies is complicated for various reasons. All are imperfect as cancer detection tools and thus are only part of a multipronged diagnostic strategy, which may include invasive procedures such as biopsy. However, given the physical and psychological consequences of biopsy, radiologists, attending physicians, and patients all have varying attitudes regarding the thresholds for follow-up and biopsy. Furthermore, factors such as study population (e.g., age distribution, types and subtlety of lesions, and a screening versus a diagnostic population), reader variability, and technical considerations (e.g., positioning of the breast and display adjustment) can greatly affect the observed performance of a device. Only a study with a very large sample size could control for all these factors, and even then, distinguishing the performance of a device from that of the reader or technologist could be quite difficult.
CASE STUDIES OF FDA APPROVAL
Numerous breast cancer detection modalities have been reviewed or are under review by FDA. These include palpation aids, mammography, CAD, ultrasound, electrical impedance, scintimammography, MRI, thermography, infrared imaging, biopsy techniques, and ductal lavage (see Chapters 1 to 3). To take a closer look at the FDA approval process for breast cancer detection devices, the remainder of this chapter examines several case studies in greater detail.
Digital Mammography
Following passage of the Medical Device Amendments of 1976, mammography devices were classified into the Class II category along with
Page 157
most other standard X-ray devices that were already in use. As a result, the clinical efficacy of these devices was essentially “grandfathered” for approval. The required 510(k) reviews for film-screen devices looked only at the technological characteristics of the mammography systems and examined some sample films to ensure that they were “substantially equivalent to” (as good or better than) the pre-1976 devices. Data on the sensitivity and specificity of FSM devices were not required.
However, when full-field digital mammography (FDDM) came onto the scene, FDA took a very different approach to evaluate the safety and effectiveness of the device and to establish its equivalence with standard analog mammography (Table 4-8). In 1995, the Radiological Devices Panel of FDA held its first meeting on digital mammography. At that time, the panel decided that diagnostic comparisons between film-screen and digital mammography (an agreement study) would be sufficient to establish the substantial equivalence of FFDM to FSM, thus avoiding large screening trials, which would be both time-consuming and costly. A guidance document was issued in 1996, in which the agency suggested use of the 510(k) clearance process. Consequently, between 1996 and 1998, the manufacturers undertook multi-institutional studies, each with 500 to 800 women who were scheduled for diagnostic mammography due to a suspicious finding on FSM or a palpable abnormality. The women underwent digital mammography, and the results were compared with those on the original mammogram obtained by FSM. Unfortunately, such a design loaded the test set with both true-positive and false-positive filmscreen mammography results, and thus, the results were biased toward FSM in terms of sensitivity and toward FFDM in terms of specificity (Lewin, 1999), making a valid comparison of these measures impossible.
The substantial equivalence of FFDM and FSM could also be established by either comparing the percentage of matches between FFDM and FSM results or looking for agreement between groups of FFDM readers and FSM readers. However, the variability among multiple readings of mammograms is well documented (Beam et al., 1996; Elmore et al., 1994; Thurfjell et al., 1994), and this inherent variability was in fact large enough to obscure any differences between FSM and FFDM readings, thus making the results difficult or impossible to interpret. The other problem with an agreement study is that even if a new technology is superior to the current standard, it would still fail the equivalency requirement because the difference in performance would be considered nonagreement. The Radiological Devices Panel met again in 1998 to discuss alternative study design options, but the members of the panel could not reach a consensus on how to proceed, so in September 1999, the agency issued letters to various FFDM sponsors requesting that they each discuss individual applications with FDA. The letter also suggested alternative pathways to approval, including the PMA application process.
Page 158
|
Date |
Event |
Summary |
|
1976 |
Medical Device Amendments |
Mammography and other X-ray devices already in use are classified as “Class II.” Pre-1976 systems receive “grandfathered” approval. After 1976, systems require 510(k) clearance or PMA application approval. |
|
1995 |
First FDA Radiological Devices Panel meeting on digital mammography |
Panel decided that to avoid costly and time-consuming large screening trials, diagnostic comparisons (an agreement study) between FSM and FFDM would be enough to establish equivalence. |
|
1996 |
Publication of FDA guidance document |
FDA suggests the 510(k) approval process. |
|
1996–1998 |
Multi-institutional studies undertaken by sponsors |
In response to the FDA guidance document for 510(k) clearance, manufacturers conduct comparison studies with 500– 800 women with suspicious findings. Test design results in FSM sensitivity bias and FFDM specificity bias. Valid comparison essentially impossible. Variability among multiple readings is also too great to compare diagnostic matches between FSM and FFDM. |
|
1998 |
Second FDA Radiological Devices Panel meeting on digital mammography |
Discusses need for alternative study design options. No consensus on how to proceed. |
|
Sept. 1999 |
FDA issues letter to sponsors |
Letter sent to various FFDM sponsors requesting separate meetings to discuss individual applications. Letter suggested alternative ways to approval including the PMA application process. |
|
Dec. 1999 |
Third FDA Radiological Devices Panel meeting on digital mammography |
FDA looks at the first individual PMA application: for the General Electric Senographe 2000 D. |
|
Jan. 2000 |
FDA grants General Electric premarket approval, with conditions |
General Electric Senographe 2000 D receives premarket approval for printed film (hard-copy) use. |
|
Nov. 2000 |
FDA approves General Electric Senographe 2000 D for softcopy use |
No other companies' digital mammography systems have been approved to date. |
Page 159
In January 2000, General Electric's machine, the Senographe 2000 D, was approved for hard-copy use (i.e., with printed film) through the PMA application mechanism. General Electric examined a diagnostic population, as in the original studies, to keep the number of women tested to a minimum. However, in this case the test population was assessed in a screening context, in which the women underwent both FFDM and FSM at the same time and several radiologists read the results on both new mammograms. This approach ensured that the number of cancers detected in the “screen” would be larger than the number detected in a general screening population. The results of the study with approximately 650 women showed that the sensitivity and specificity of FFDM lie within an acceptable range of the values calculated for FSM (i.e., less than a 5 percent difference). The approval order did not specify any post-approval studies, but it did call for expedited approval of the soft-copy modality, which was granted in November 2000. Other manufacturers are gathering data for similar submissions, but to date, none has been successful.
The approval process for digital mammography was complicated, lengthy, and very costly for the developers. Critics have questioned whether a PMA application or extensive clinical data were really necessary, given that the technology of x-ray interrogation of the breast in FFDM is identical to that in FSM, interpretation techniques are similar, and the efficacy of mammography has already been
established.22
When digital detectors for chest X rays were approved, the devices underwent a vastly simpler 510(k) clearance process with little consideration of how the diagnostic images generated would be used or interpreted.23
Other breast imaging technologies, such as breast MRI and thermography, have also been cleared by this approach. In the case of breast MRI, only the breast coil had to be cleared by the 510(k) process by demonstrating substantial equivalence with MRI devices used to image other parts of the body.24
No consideration was given to the accuracy of interpreting the images generated by the breast coil. Similarly, the BioScan system, a thermal imaging device manufactured by OmniCorder, was cleared by the 510(k) process for use as a diagnostic adjunct for breast cancer detection.25
The device was deemed equivalent to other thermal imaging tech-
22Radiological Devices Panel Meeting Transcript, December 16, 1999.
23Chest X rays are not used for screening, and diagnostic chest X rays are generally less difficult to interpret than mammograms.
24MRI devices were originally labeled as Class III devices because safety issues with regard to exposures to strong magnetic fields were undefined. More recently, MRI devices were downgraded to Class II.
25Several 510(k) applications for thermal imaging devices have been cleared since 1976 for use as adjunct technologies for the diagnosis of breast cancer.
Page 160
nologies, although none of those technologies had been proved to be valuable for the detection of breast cancer.
According to FDA, however, the inherent “risk” of digital mammography lay in the fact that millions of women rely on mammography for early detection of breast cancer, with implications ranging from breast conservation to saved lives. Thus, in assessing FFDM, FDA stated that it wanted to ensure that the new technology would improve on the successes of FSM or, at the very least, that no loss in sensitivity and no clinically important loss in specificity would occur (usually, FDA requires demonstration of substantial equivalence, not the superiority of new devices). In other words, FDA wanted companies to demonstrate that FFDM would not miss more cancers than the current technology and that the number of unnecessary biopsies in women without cancer would not increase.26 The ease of handling information acquired digitally was not deemed, in itself, sufficient to support marketing of FFDM unless these two clinical objectives could be met.
The FDA policy and requirements for approval changed multiple times during the approval process. Throughout the discussions the advisory panel struggled with questions of whether digital mammography qualified as a “new” technology and how to define “safety and effectiveness.” On the one hand, digital mammography exposes women to a similar level of ionizing radiation as analog film mammography, thus posing no additional direct physical harm to the woman. The difference between FFDM and FSM is the acquisition and display of the images. Whether this significantly changes the “safety and effectiveness” of mammography subsequently determines which approval process [i.e., the 510(k) process versus the PMA application process] is required. In the end, the panel decided to let individual companies decide on their own particular course, with FDA offering guidance. The PMA application process was offered as an alternative because it allows greater flexibility in the study requirements, permitting some questions to be addressed later in the post-marketing period.
In summary, FDA was faced with a challenging evaluation and had good intentions—protecting American women from false-positive findings during breast cancer screening—when it issued its guidance documents for the approval of digital mammography. However, in the end, following those guidance documents led to significant delays in the approval of FFDM, at great expense to the sponsoring companies. The difficulties encountered in attempting to demonstrate the substantial clinical
26Transcript from an FDA panel meeting
(http://www.fda.gov/ohrms/dockets/ac/98/transcpt/3446t2.rtf).
Page 161
equivalence of FFDM to FSM could have been predicted, given the well-known variability in the interpretation of mammograms. Thus, the initial expectations of FDA for the clinical studies were unrealistic, and indecision on the part of FDA following that realization further contributed to delays in approval.
Pro*Duct Breast Catheter
The Pro*Duct catheter (Love et al., 2000) is an example of a device that could potentially be used for breast imaging (contrast-enhanced radiography) as well as for biological tests (ductal lavage for collection of breast fluids and cells for analysis). The catheter was recently cleared by FDA via the 510(k) process, with no assessment of how it would be used for breast cancer screening or diagnosis. Clearance was granted on the basis of the indication that it “enables the collection of breast milk duct fluid for cytological evaluation.”27 The product label further stipulates that the “collected fluid can be used in the determination and/or differentiation of normal versus premalignant versus malignant cells.” The label specifies that the device should be used only as an “adjunct to standard breast cancer detection methods, including mammography and physical exam.” However, the company has not conducted clinical trials to determine the sensitivity and specificity of this technique and has not compared it with other screening and diagnostic methods. Thus, FDA required a precautionary statement in the label to indicate that “sensitivity/specificity data for ductal cytology from well controlled clinical trials is not currently available. The use of the information in clinical practice must therefore be determined on a case by case basis.” No guidelines as to how to make such decisions are provided. Furthermore, it is not clear what action should be taken if, in the absence of a mammographic finding, the results obtained by the procedure indicate a malignant or premalignant lesion.
TransScan Electrical Impedance Imaging
TransScan Medical (Ramsey, New Jersey) initially sought FDA premarket approval in 1997 for its electrical impedance imaging device, the T-Scan 2000 device, as an adjunct to mammography for women with indeterminate lesions. Preliminary discussions with FDA and protocol reviews began as early as mid-1994, and in 1997 the company submitted data from a large, multicenter study with a screening population imaged
27 Angela B. Soito, vice president of regulatory and quality affairs, Pro*Duct Health, Inc., personal communication, September 2000.
Page 162
both by mammography and with the T-Scan 2000 device. The mammogram and impedance image for each woman were read blindly. The device was not intended to be used for screening, but it was thought that a double-blind study with a screening population would be the most rigorous and unbiased way to evaluate the device. In this setting, the company reported that for women with equivocal mammograms, the adjunctive use of the T-Scan 2000 device improved the sensitivity of mammography by 22 percent and the specificity of mammography by 16 percent. Although the FDA advisory panel considered the device to be promising, it expressed concerns about how it would be used in practice and whether its use would increase the number of biopsies or cause some women with cancer to forego biopsy. As a result, approval of the T-Scan 2000 device was denied, and the FDA panel recommended that the company identify the population that would benefit most from the device and conduct more studies targeted at that population.28
TransScan sought FDA premarket approval again in 1998, and in April 1999 the device was approved as an adjunct to mammography for women with equivocal lesions in BIRADS29 Categories 3 and 4. TransScan submitted data from two additional studies conducted with the targeted population and under conditions more closely resembling those for its intended clinical use. Statistical modeling was used to combine the results of those studies with the results of the original double-blind study. The combined results indicated that the T-Scan 2000 device, when used in conjunction with mammography on a targeted population, improved the diagnostic sensitivity by 15.6 percent and the diagnostic specificity by 20.2 percent over those from the use of mammography alone. The device is not to be used for the assessment of lesions with clear mammographic or nonmammographic indications for biopsy.
TransScan is conducting additional studies to further validate the technology, as required by FDA as part of its premarket approval process. Post-approval studies must look at the clinical use of the T-Scan 2000 device and any consequent changes in sensitivity and specificity, as well as the effects of the menstrual cycle on the performance of the device.30
28Radiological Devices Panel Meeting, Center for Devices and Radiological Health, FDA, November 17, 1997
(http://www.fda.gov/ohrms/dockets/ac/97/transcpt/3353t1.pdf).
29The Breast Imaging Reporting and Data System (BIRADS) includes five categories of assessment with increasing suspicion of malignancy, along with standard follow-up recommendations for each category. Category 3 is “probably benign,” whereas Category 4 is “suspicious abnormality.” For more detail, see
Chapter 1.
30John Neugebauer, vice president of marketing, TransScan Medical, personal communication, [June, 2000].
Page 163
Computer-Aided Detection
CAD was developed with the intent of reducing observational error in the reading of mammograms. CAD systems identify and mark regions of interest in screening mammograms, thus assisting radiologists in locating areas that might warrant closer inspection. In November 1996, the company R2 Technology, Inc., began discussions with FDA regarding clinical issues and protocols that would be appropriate to determine the safety and effectiveness of its M1000 ImageChecker system. As a result of those discussions, three clinical studies with screening populations were designed for premarket approval. Those studies concluded that (1) CAD does not increase the diagnostic workup rate; (2) CAD correctly marks a high percentage of microcalcifications and masses, thus minimizing the rate of false-negative results; and (3) the system generates reproducible results with good sensitivity for the identification of microcalcifications and masses associated with cancer. The Radiological Devices Panel reviewed the application in May 1998, and in June 1998, R2 Technology was granted premarket approval of its M1000 ImageChecker. FDA approved the technology on the condition that a post-approval study be performed to more accurately assess the effect of the device on the rates of true-positive and false-negative results.
In its PMA application, R2 Technology did not list any direct risks to health or safety; however, as an indirect risk, it did mention missed lesions and false-positive results as potential adverse effects. The company does not market the device as a replacement for the radiologist; rather, it is the combined efforts of the radiologist and the device that result in the increased sensitivity. Since there were no direct physical safety issues, the question of effectiveness focused on the radiologist-device combination. In fact, this issue is pertinent for most screening and diagnostic devices, since human interpretation is an essential component of the detection process. The question then becomes whether the approval process should focus solely on the physical device or whether the interpretation and potential consequences of that interpretation should be considered.
SUMMARY
Over the last 10 years, efforts directed toward the development of new technologies for early breast cancer detection have increased significantly. Many new funding initiatives have been launched, some of which are the direct result of consumer advocacy, and new collaborative efforts have been undertaken. Government funding in particular has recently placed a new emphasis on the translation of science through the development of technology, in contrast to the more traditional focus on basic scientific discovery. Notably, there has been an increase in joint public
Page 164
sector-private sector efforts. This increase in funding opportunities could stimulate progress in the development of new detection technologies.
Private-sector investment in medical imaging technologies is considerably less than private-sector investment in other areas of the health care industry, perhaps because of the perception of risk associated with such ventures. The technology development process in general is complex and costly, and the end results of research are unpredictable, making it a financially risky undertaking. For medical devices, the FDA approval process adds an additional level of risk to the development process (as can coverage decisions, which will be discussed in the next chapter). The requirements for approval have been variable and unpredictable in the past, in some cases increasing the cost of obtaining approval and the length of time needed to enter the market. The subsequent lag in development can conceivably devastate companies that are funded to produce a return on investment in a relatively short time.
Regulation of medical device manufacturers is clearly necessary and beneficial, but also quite challenging. Without regulation, the market could potentially be flooded with unsafe devices. The drawback is that the regulation increases costs and can slow the process of technology release. Problems can arise if decisions regarding regulatory requirements are inconsistent, unclear, delayed, or faulty. For example, assigning technologies to a particular approval pathway [the 510(k) process versus the PMA application process) can have enormous ramifications for the sponsors in terms of the cost of obtaining approval, but these designations have not always been made in a consistent fashion. Similarly, different operational definitions of “substantial equivalence” could have a significant effect on the process of how equivalence is determined. In the case of FFDM, these definitions and designations changed throughout the approval process. Thus, the approval system could be improved by establishing and more clearly defining a requisite level of agreement between technologies and a required level of statistical precision. Given the numerous complexities in assessing new technologies, FDA advisory panels would benefit from the addition of more experts in biostatistics, technology assessment, and clinical epidemiology.
Separate study guidelines for diagnostic and screening purposes are necessary, including delineation of appropriate end points and study sizes. The dominant framework for medical technology development and evaluation has historically been based on therapeutics, whereas early detection relies on screening and diagnostic methods. The evaluation of such methods may be intrinsically different. The “safety” assessment of screening and diagnostic devices examines direct patient harm from use of the device (analogous to therapeutics), but can also include indirect harm from unnecessary interventions that are pursued on the basis of information generated by the device. Similarly, evaluation of the “effec-
Page 165
tiveness” of screening and diagnostic tests is fundamentally an assessment of the value of the information obtained from the test, in contrast to the direct effects of therapeutics on clinical outcome. Most patient-level effects of these devices are mediated by subsequent therapeutic decisions.
As a result, there are difficulties associated with comparing new technologies to the imperfect “gold standard” of FSM. The intrinsic variability in the production and interpretation of mammograms and other breast images makes it difficult to accurately determine the sensitivity and specificity of imaging modalities. This variability caused major difficulties and delays in the FDA approval process for FFDM, and thus greatly increased the cost and time required to gain approval.
Using measures of accuracy (e.g., sensitivity and specificity) to make approval decisions may be appropriate for most diagnostic devices. In the case of “next-generation” devices (in which technical improvements have been made to a predicate device already on the market), technical advantages such as patient comfort or ease of data acquisition and storage could be considered in determining approval as well. Ideally, all new screening technologies would be compared to gold standard technologies by using the reduction in disease-specific mortality as an end point. However, such an approach is logistically impractical for device companies because of the study size, length, and cost. A more coordinated approach for the testing of new screening technologies, with input and support from FDA, NCI, health insurers, and patient advocates may help to overcome this barrier. If a new device approved for diagnostic use shows potential for screening use and the developers wish to pursue approval of the device for screening, an investigational device exemption should be granted for this use based on measures of accuracy. In addition, conditional coverage (as discussed in Chapter 5) could be provided for the purpose of conducting large-scale screening trials to assess clinical outcomes. This approach would prevent “off-label” adoption of detection technologies that have not been assessed in the screening setting. The recently launched ACRIN trial for the study of digital mammography may provide an example of how screening studies could be conducted under this proposed mechanism.
FDA approval is only the first hurdle that new technologies face once they have been developed. Although both public and physician perception is that FDA approval means that technologies “work” and reimbursement for use of the device should be provided, coverage decisions are rarely that straightforward. For example, FDA approval does not mean that a new device is better than its predecessor or that it is useful for applications for which it has not been evaluated. Approval simply allows a company to sell the device, but ultimately, coverage decisions by third-party payers are likely to determine whether the technology becomes widely used and disseminated. This issue will be examined more closely in Chapter 5.


































