
GERHARD LUDWIG CLOSS
May 1, 1928–May 24, 1992
BY HEINZ D. ROTH
WHEN GERHARD LUDWIG CLOSS succumbed to a massive heart attack on May 24, 1992, the work of one of the great organic chemists came to an untimely end. Professor Closs made significant contributions in four areas. He was an early leader in the field of carbene chemistry; he elaborated various significant aspects of the photosynthetic pigments; he pioneered important applications of magnetic resonance to characterize reaction intermediates; and he elucidated intricate facets of electron transfer chemistry. This biographical memoir provides a welcome opportunity to pay tribute to one of the outstanding chemists of the post-World War II era—and to a friend.
Gerhard Closs was born on May 1, 1928, in Wuppertal-Elberfeld, Germany, a small, bustling city known for its suspended tram (Schwebebahn) and for the pharmaceutical branch of Bayer, one of Germany’s major chemical manufacturers. Before he could complete his high-school education he was pressed into military service as a 16-year-old in 1944. He barely survived the ordeal of war: He was seriously wounded on the eastern front. After the war he completed high school and enrolled in Universität Tübingen. Having received a Ph.D. degree in 1955 for work with Georg Wittig, he joined R. B. Woodward’s group at Harvard for two years.
In 1957 he accepted a position as assistant professor at the University of Chicago as a natural products chemist.
Gerhard’s early independent studies were assisted by Lieselotte E. Closs (née Pohmer), his wife and most productive coworker; their collaboration produced 15 publications between 1959 and 1969. Lieselotte also received a Ph.D. degree with Wittig for her classic work on the transient existence of dehydrobenzene.1,2 She did postdoctoral work at MIT. Lieselotte and Gerhard were married on August 17, 1956, in Cambridge, Massachusetts.
Employment opportunities for women scientists were very limited in the 1950s and 1960s. University regulations against nepotism prohibited wives from holding paid positions in the same department as their husbands; therefore, Lieselotte could only work as an unpaid volunteer. The availability of a skilled coworker proved especially fortuitous when Gerhard entered the chemically induced dynamic nuclear polarization field (see below). Lieselotte re-entered the lab, carried out a few simple but elegant experiments to probe key aspects, and soon had results sufficient for two “Communications to the Editor.”
Gerhard Closs was granted tenure in 1961 and was promoted to full professor just two years later. Almost 20 years later he accepted the position of section head in the Chemistry Division at Argonne National Laboratory, while remaining on the Chicago faculty. Although he kept this position for only three years, it significantly influenced the direction of his research in the final decade of his life.
The work of Gerhard Closs has been recognized at the University of Chicago and in the scientific community at large. He was appointed the Michelson Distinguished Service Professor, and his colleagues honored him along with N. C. Yang with a symposium on the occasion of their sixti-
eth birthdays. He was awarded the Jean Servas Stas Medal by the Belgian Chemical Society in 1971, the James Flack Norris and A. C. Cope awards by the American Chemical Society in 1974 and 1991, respectively, and the Photochemistry Prize by the Inter-American Photochemical Association in 1992. He was elected a member of the National Academy of Sciences in 1974 and the American Academy of Arts and Sciences the following year. The Inter-American Photochemical Association honors his memory with the G. L. Closs Memorial Award, which allows a student to present a research paper at one of its meetings.
In 1981 he also was honored as chairman of the Gordon Research Conference on free radical reactions and in 1990 the Gordon Research Conference on radical ions, and by many distinguished lectureships in the United States, Canada, Japan, and Europe. Among these were the Bayer Lectureship at Universität Köln, Germany (where the author first met him), a visiting professorship at Yale (where the acquaintance was renewed), regular visits to Bell Laboratories, and the Merck Distinguished Lectureship at Rutgers University (where the author was privileged to be his host).
Closs was a featured speaker at many national and international congresses and symposia, and his participation at meetings was a highly important contribution to science. He brought to these meetings a keen analytical mind and the command of an unequalled breadth of chemical topics: from subtle details of organic synthesis, to a deep understanding of mechanistic details, to the intricacies of chemical physics, and a keen chemical intuition. This combination allowed him to probe proposed theories or mechanisms as they were being presented. Few of his peers made more pertinent comments than Gerhard did, or in a more impertinent fashion when he felt it necessary. Even accomplished
and experienced lecturers must have felt a tinge of apprehension when he raised his hand and, on being recognized, uttered his familiar, “I would like to take issue with …”.
Gerhard Closs relaxed by sailing, and sometimes racing, his sailboat on Lake Michigan for hours, days, or weeks. He relished his fine collection of graphic art, he was stimulated by theater performances, including modern and avantgarde plays, and he enjoyed classical music. No matter how hard the author tried, however, Gerhard could not be persuaded to attend an opera performance. (He had sworn off opera as a teen in the early 1940s, following a performance of Da Ponte and Mozart’s Cosi Fan Tutti in Wuppertal.)
During his 35 years at the University of Chicago, Gerhard developed a deep appreciation, even love, for his adopted country. He only bought American cars and “took issue” with many Americans and foreigners alike who dared to criticize the United States in his presence. One afternoon, while attending a Gordon Conference in New Hampshire, Gerhard was interviewed by a local reporter. Asked what he thought of consumers who bought foreign-made articles, he quickly voiced his disapproval and then added, having spotted the reporter’s Japanese-made camera, “and that applies also to you.”
With the death of Gerhard Closs the chemical sciences lost a most formidable champion, a practitioner of the highest intellectual standards, a keen mind, and a skilled experimenter who was always probing accepted theories and was never afraid to break new ground. The scientific community has lost a teacher, mentor, collaborator, and kin spirit, and a few who were privileged have lost a friend.
The earliest publications of Gerhard Closs stem from his thesis work with Wittig and describe ylid rearrangements with ring enlargement or contraction,3 and from his postdoctoral training with R. B. Woodward (total synthesis
of chlorophyll).4 His first independent publications, for example, a paper on the active constituents of Panaeolus venenosus,5 reflect his being hired as a natural products chemist. As part of the venenosus project the physiological effects of the mushroom were to be tested, and the young assistant professor volunteered for the study. The highly amusing conversation ensuing between Gerhard Closs and his physician and collaborator is part of the Mycologia publication.5 It was often cited at Closs group festivities and never failed to amuse; coworkers fortunate to have obtained a reprint of this paper count it among their prized possessions.
Gerhard Closs never lost interest in natural products and photosynthetic pigments. Seventeen of his 132 lifetime publications dealt with the chlorophylls; he contributed significantly to such important topics as linked chlorophyll dimers, photosynthetic reaction centers, and porphyrin metal complexes. Still, his most significant contributions came in three other fields of chemistry, one area for each decade of his professional career.
Gerhard Closs’s first major contributions came in the emerging field of carbene chemistry; interestingly, a distant predecessor at the University of Chicago, John U. Nef, was an early champion of divalent carbon chemistry. Alas, Nef’s interpretations of his results are at variance with the accepted definitions and the prevailing understanding in the field since the mid-twentieth century so that his work no longer qualifies as carbene chemistry.6
The actual roots of the carbene field lie in the basecatalyzed hydrolysis of trichloromethane by Geuther in 1862.7 Hine repeated this experiment in 1949 and recognized the reaction as an α-elimination, the consecutive removal of H+ and Cl– from the same carbon, generating dichlorocarbene.8 In 1954 Doering and Hoffman trapped the postulated spe-
cies by addition to cyclohexene, demonstrating its intermolecular reactivity.9
(eq.1)
(eq.2)

(eq.3)
The development of divalent carbon chemistry involved various facets: substituted carbenes; the notion of spin multiplicity; chemical studies probing carbene reactivity and the stereochemistry of their reactions; and the application of new physical methods (e.g., electron spin resonance, electron nuclear double resonance, chemically induced dynamic nuclear polarization, and optical spectroscopy). Gerhard Closs played a significant role in introducing these new techniques to the study of carbenes.
Closs generated chlorocarbene from methylene chloride; addition of the new carbene to alkenes, benzene, or phenol gave rise to chlorocyclopropanes,10 tropylium chloride,11 or tropone,12 respectively. Five-membered heterocycles (e.g., pyrrole and indole) reacted with chlorocarbene by ring expansion.13 It is tempting to see in these ring enlargements echoes of his doctoral thesis.

Additional carbenes arose by reaction of alkyl and benzal halides with organolithium compounds; here the term “carbenoid” was introduced to denote carbenes that appeared to be complexed (i. e., associated) with lithium halide.14 The base-induced α-elimination of chloroalkenes formed cyclopropenes by intramolecular addition of alkenylcarbenes.15
By the time of his promotion to associate professor he began to ask further-reaching questions; he decided to characterize carbenes more thoroughly and, if possible, observe them directly. Spectroscopic techniques available at this time included optical spectroscopy and electron spin resonance. Optical spectroscopy had received a recent boost by the advent of flash photolysis in 1949-50.16 Herzberg observed the emission spectra of the parent methylene, CH2, and its isotopomers CHD and CD2, in 1961.17
Electron spin resonance (ESR) spectroscopy was a later development,18 but by 1953 organic free radicals or radical ions had been studied. Gerhard Closs was fortunate to have Clyde Hutchison, an expert ESR spectroscopist, as a colleague. They generated diphenylmethylene at cryogenic temperatures in benzophenone crystals and observed the first ESR spectrum of a ground state triplet carbene in a single crystal.19 About two weeks before the Chicago group, Edel Wasserman and coworkers at Bell Laboratories generated diphenylmethylene in a glassy matrix.20 The single crystal approach of the Chicago collaborators lent itself to a more detailed analysis and interpretation. Ultimately, electron nuclear double resonance (ENDOR) revealed the detailed structure of this intermediate (see Figure 1).21 Among Closs’s additional ESR studies cyclopentanediyl and trimethylcyclopropenyl deserve special mention.

FIGURE 1 Structure of diphenylmethylene as derived from ENDOR experiments.21
Following the work on the ESR spectroscopy of triplet states Gerhard Closs studied triplet carbenes by optical spectroscopy,22 including the pioneering time-resolved laser spectroscopy study of diphenylmethylene.25 Two other groups probed optical spectroscopy of diphenylmethylene independently,23,24 and only one study had dealt with the application of time-resolved laser spectroscopy (TRLS) to carbene chemistry26 when Closs and Rabinow’s study of diphenylmethylene addition to alkenes25 opened the field to studies in other laboratories. Although the limited (μs) time resolution of these early studies appears almost primitive compared to today’s sophisticated TRLS experiments, the available time resolution was exactly right for the somewhat “sluggish” diphenyl-methylene.
Related studies involved cyclopropenes and bicyclobutanes, newly accessible with his new carbenes, notably the isomerization of 2,4-dimethylbicyclo[1.1.0]butane to butadiene. The conservation of orbital symmetry, a concept developed by Woodward and Hoffmann in the early 1960s,27
predicts that this reaction will proceed as a concerted [σ2s+σ2a] process with predictable stereochemistry for the migrating carbon centers. Closs and Pfeffer probed the rearrangement of two 2,4-dimethylbicyclobutanes to two hexadienes and elucidated the steric course of this reaction.28

By the mid-1960s Gerhard Closs, an acknowledged expert in carbene chemistry, made his final major contribution to this field, the first application of the chemically induced dynamic nuclear polarization method. In 1967 enhanced nuclear magnetic resonance (NMR) emission was observed in some chemical reactions,29,30 yet another facet in the rich palette of NMR applications. Because some reactions giving rise to NMR emission were known free-radical reactions, these effects were explained as electron-nuclear cross relaxation, hence the designation “chemically induced dynamic nuclear polarization” (CIDNP) for the new phenomenon. However, this mechanism was soon found wanting, as an increasing number of effects were incompatible with the cross relaxation mechanism.
Gerhard Closs immediately recognized the value of this technique. With his thorough understanding of organic reaction mechanisms and his expertise in the physical principles underlying magnetic resonance, he was in a unique
position to elucidate the physical and chemical principles underlying CIDNP. He entered the field with all his vigor. He persuaded Lieselotte to return to the bench for a few well-designed experiments.31,32 In 1969 four back-to-back communications appeared in the Journal of the American Chemical Society, followed quickly by six more, for a total of ten communications in only 20 months. After two CIDNP studies in photoreactions of diphenyldiazomethane31 and benzophenone32 Closs began to probe the actual origin of the spin polarization effects.
Recognizing that all CIDNP effects required the involvement of radical pairs,33 he developed a theory that could explain the observed polarization and designed elegant experiments to probe key features of the theory. Because the polarization changed with the spin multiplicity (μ) of the precursor from which the pair was generated, he suggested CIDNP “as a tool for determination of spin multiplicities of radical pair precursors.”34
He also recognized that the polarization pattern of CIDNP effects depends critically on the relative g factors (Δg) of the paired radicals and illustrated the effect of systematical changes in the g factor difference (Δg) between the interacting radicals on the CIDNP spectra (see Figure 2).35

In this context I will share an anecdote about the 1970 American Chemical Society meeting in Houston, Texas, where
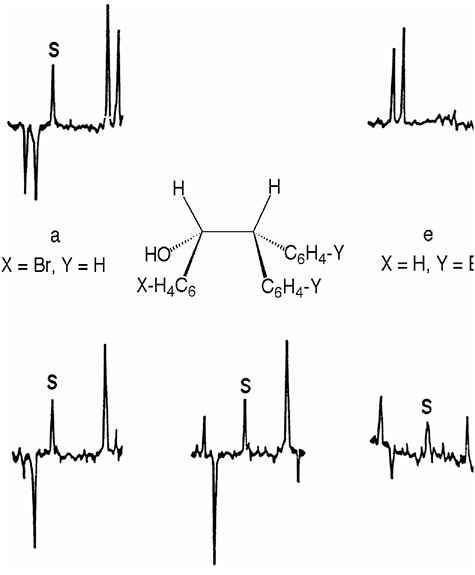
FIGURE 2 CIDNP spectra (benzylic protons) of coupling products generated during the photoreactions of benzaldehyde (X = H) and derivatives (X = Cl, Br) with diarylmethanes (Y = H, Cl, Br).33
Gerhard first disclosed major features of the proposed theory. The ACS Organic Division sponsored a symposium about the new phenomenon; most of the early contributors were present, except the Japanese and Russians. Gerhard was scheduled to give the opening lecture of the afternoon session. After brief introductory remarks he asked for the first slide, studied it briefly, asked quickly for the second and third slides, then turning to the session chairman, said, “Mr. Chairman, I know this is highly irregular; I thought this could only happen to Michael Dewar [Professor Michael Dewar, for four years, 1959-63, Closs’s colleague at the University of Chicago; University of Texas, Austin 1963–]. I took the wrong set of slides.” The next speaker was asked to present his talk out of turn, and Gerhard retrieved the correct set of slides from his hotel, which fortunately was just across the street. When he returned, he showed a series of astonishing slides that were well worth the (brief) wait (as well as the violation of the ACS guidelines for the presentation of papers).
Coincidental with the Chicago group, L. Oosterhoff and R. Kaptein at Universiteit Leiden (Netherlands) worked on the mechanism of CIDNP and demonstrated additional elements of the theory. They noted that in-cage products and cage-escape (free-radical) products showed polarization of opposite sign,36 and that protons with hyperfine coupling constants of opposite sign show CIDNP effects of opposite sign.36 The Radical Pair Theory emerging from the work of the Chicago and Leiden groups is now generally accepted and can explain the vast majority of all nuclear spin polarization effects.
In more than 20 additional publications Closs and collaborators elucidated additional facets of the theory of CIDNP or dealt with applications to new problems. They made ma-
jor contributions to understanding biradicals: The magnetic field dependence of CIDNP effects of biradicals yields their average singlet-triplet splitting,37 as well as their lifetimes (see Figure 3).38 Closs and colleagues also established relaxation39 and cross relaxation phenomena.40 Cross relaxation transfers nuclear spin polarization to nuclei, which are not coupled to an unpaired electron spin. The existence of cross relaxation complicates the interpretation of CIDNP results, particularly in experiments designed to derive electronic structures of free radicals or radical ions from their polarization pattern.
Closs also pioneered flash photolysis with CIDNP detection, combining the advantages of TRLS with the structural
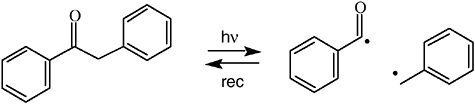
information content unique to CIDNP.41-43 “The basis for the success of the time-resolved method is the fact that geminate processes are complete in a fraction of a microsecond, while combination of free ions and/or exchange … may take tens or hundreds of microseconds depending on concentration.”42 This method was soon adopted in other laboratories for various applications. The Closs group applied this technique to determine the spin density distribution in radical ions of chlorophyll and derivatives. The CIDNP intensities 1 μs after a photoinduced electron transfer reaction revealed the signs and relative magnitudes of hyperfine coupling constants43 in good agreement with ENDOR spectroscopy results. Another application probed the kinetics of triplet states and biradicals; photolysis of deoxybenzoin cleaves a C–C bond next to the carbonyl group. The resulting radical pair can regenerate the reactant by geminate recombination; free radicals escaping from the geminate cage may have secondary encounters, forming deoxybenzoin or bibenzyl. At the shortest delay then attainable (1 μs) the reactant showed the expected geminate polarization; as expected, when the delay time was increased to 100 μs, this signal grew, due to contributions from free radical combination (see Figure 4).41
Without any doubt Gerhard Closs’s contributions have made the CIDNP method a well-defined, sophisticated, powerful, and reliable technique with a wide spectrum of important applications. When he first came to Chicago, super-
vision of the departmental NMR instrument was one of the tasks assigned to him. Surely Gerhard Closs took this responsibility seriously and made the most of the opportunities this position offered.
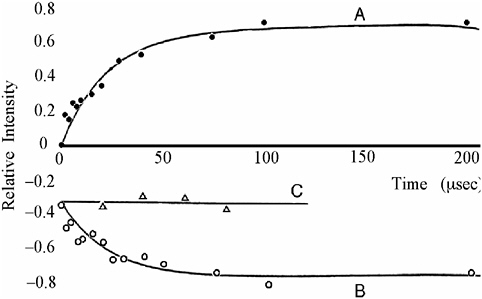
FIGURE 4 Normalized line intensities of bibenzyl (A) and deoxybenzoin (B), observed during the photolysis of deoxybenzoin, as a function of delay time T. The deoxybenzoin polarization in the presence of a (thiol) free radical scavenger is designated C.41
In the final decade of his life Gerhard Closs became interested in several aspects of chemically induced dynamic electron polarization (CIDEP), a phenomenon related to CIDNP and discovered several years earlier.44 Essentially all CIDEP effects observed in the 20 years following the discovery could be explained by the radical pair or the triplet mechanism or by a combination of both. In the mid-l980s several time-resolved CIDEP spectra with highly unusual fea-
tures were observed upon photolysis of ketones in micelles,45 which could not be explained by the existing theories. As a natural extension of his earlier work on biradicals and in the context of his interest in the superexchange mechanism of intramolecular electron transfer (see below), Gerhard Closs investigated related systems in collaboration with M. Forbes and J. Norris. They recognized the unusual effects as manifestations of electron spin-spin interactions; the resulting effects are rapidly lost in solution but they “remain observable because of limited diffusion in micelles.”46 McLauchlan and coworkers derived a similar explanation independently.47 The concept of spin-correlated radical pairs is now generally accepted.
Closs’s time-resolved electron spin resonance studies culminated in several elegant studies of electron spin polarized polymethylene biradicals in solution, in which through-bond interactions were illuminated.48 Simulating the shape and time dependence of CIDEP spectra from acyl-alkyl and alkyl-alkyl polymethylene biradicals by perturbation theory yielded the electron spin-spin interaction, J, and the end-to-end contact rate, which is inversely related to the lifetime. It is remarkable that Gerhard Closs was still breaking new ground at the onset of his seventh decade.
In 1979 Gerhard Closs accepted the position of section head in the Chemistry Division at Argonne National Laboratory. He kept this position for only three years, but this appointment significantly influenced the direction of his research in the final decade of his life. He took an interest in the electron transfer work of John Miller, who had evidence for the “inverted Marcus region.” More than three decades earlier Marcus formulated the rate of an electron transfer reaction as a function of two parameters, its driving force (i.e., the free energy), ΔG0, of the reaction and a
“solvent reorganization energy,” λs, required to accommodate the changing charge distribution.49
The most striking result emerging from this work was the prediction that electron transfer rates increase with increasing driving force to a maximum at λs = ΔG0, but then unexpectedly (and perhaps counterintuitively) increasing the driving force further would decrease the reaction rate. The essential predictions of this theory were reproduced by numerous theoretical approaches descending from the original idea of Marcus. It took almost 30 years before this prediction was confirmed by experiment.
Electron transfer reactions fall into three classes: charge separation, the photoinduced generation of radical ion pairs from a donor and an acceptor molecule; charge recombination, the reverse process; and electron exchange between a charged and a neutral entity. Rehm and Weller studied the charge separation in more than 60 organic donor-acceptor pairs. The rate constants of fluorescence quenching indicated a maximum rate of electron transfer, essentially diffusion limited, without evidence for an inverted region. Although at variance with the existing theories, the results were rationalized based on an ad hoc theory.50 Similar results were observed in additional systems; all attempts to verify the predicted inverted free energy dependence of electron transfer rates met with failure.
Miller and coworkers had studied the (charge neutral) electron transfer from radiolytically generated radical anions to aromatic hydrocarbons in frozen solutions with free energy changes ranging from 0.01 < –ΔG0 < 2.75 eV. The rates of electron transfer decreased at high exothermici-
ties,51 the first report of a reduction of electron transfer rates with increasing driving force. However, because the entities were randomly distributed in rigid glasses, the results were difficult to interpret and to understand.
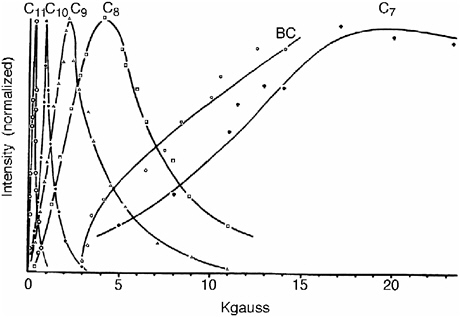
In the collaboration that ensued and continued to Gerhard Closs’s death donor and acceptor were linked by a rigid steroid spacer in molecules of the type A-Sp-biphenyl. The electron transfer rates observed for the radiolytically generated monoanions of these systems showed a striking deviation from the classical Brønsted relationship (see Figure 5),52 confirming the predictions of Marcus. Less than five months after Gerhard Closs’s death, R. A. Marcus was awarded the 1992 Nobel Prize in chemistry. In the announcement the Swedish Academy disclosed that the Nobel committee had long recognized the significance of Marcus’s theory but had waited for experimental verification before awarding the prize.

These findings stimulated major research efforts around the world. Within a year the Marcus inverted region for charge recombination was reported and elaborated; a charge separation reaction was also found to exhibit the full range of Marcus behavior. The Chicago-Argonne team continued to produce a host of significant results. They established the nonlinear dependence of electron transfer rates upon
solvent polarity, probed the distance dependence using decalin and cyclohexane spacers, and established the temperature independence of electron transfer rates in several systems. Having studied both “hole” and electron transfer rates, Closs and Miller reasoned that triplet energy transfer might be considered as the sum of the two processes. This consideration spawned an illuminating, unprecedented comparison of the rates of hole, electron, and triplet energy transfer.53 Further collaboration was cut short by his untimely death.

FIGURE 5 Intramolecular rate constants as a function of free energy change in 2-methyloxacyclopentane solution at 296 K. The electron transfer occurs from biphenyl anions to the eight different acceptor moieties (shown adjacent to the data points), in eight bifunctional molecules of the general structure shown in the center.52
One significant aspect of the electron transfer work suggested that donor and acceptor are coupled by the interactions with the orbitals of the intervening molecular fragments, a mechanism referred to as superexchange. This was one of the reasons that caused Gerhard Closs to reconsider the interaction of two unpaired electron spins in biradicals through the intervening molecular fragment, leading to the concept of spin-correlated radical pairs (see above).
Gerhard Closs made significant contributions in three fields of chemistry. He was an early leader in the field of carbene chemistry, he pioneered applications of magnetic resonance to characterize reaction intermediates, and he elucidated intricate facets of electron transfer chemistry. He will be remembered for the depth and breadth of his understanding and for his rigorous, all-encompassing approach to research. Once he became interested in a problem he would first master the theoretical background, or develop it himself, as was the case with CIDNP. Then he would identify key features that needed to be verified and design ingenious and decisive experiments to probe the theory. Finally, he would synthesize the selected target molecules and conduct the physical experiments.
IN WRITING THIS MEMOIR I have benefited from conversations with several of Gerhard’s students and colleagues, particularly in the difficult task of choosing the “Selected Bibliography” from his many superb publications: Robert A. Moss, who was a student in the carbene years (cf., 1964); John Miller, who inspired Gerhard’s interest in electron transfer and collaborated with him for his final 10 years (cf., 1983, 1984, 1986, 1989, 1990); Jim Norris, an associate and friend at the University of Chicago, who made available a tribute to Gerhard, written for the University of Chicago Chronicle;54 and Piotr Piotrowiak, one of his last students, who bridged the electron transfer and biradical projects (cf., 1989, 1992).
NOTES
SELECTED BIBLIOGRAPHY
1960 With L. E. Closs. Carbenes from alkyl halides and organolithium compounds. I. Synthesis of chlorocyclopropanes. J. Am. Chem. Soc. 82:5723-28.
1961 With L. E. Closs. Alkenylcarbenes as precursors of cyclopropenes. J. Am. Chem. Soc. 83:2015-16.
1963 With L. E. Closs. Carbon orbital hybridizations and acidity of the bicyclobutane system. J. Am. Chem. Soc. 82:2022-23.
1964 With R. A. Moss. Carbenoid formation of arylcyclopropanes from olefins, benzal bromides, and organolithium compounds and from photolysis of aryldiazomethanes. J. Am. Chem. Soc. 86:4042-53.
1965 With R. L. Brandon, C. E. Davoust, C. A. Hutchison, Jr., B. E. Kohler, and R. Silbey. Electron paramagnetic resonance spectra of the ground-state triplet diphenylmethylene and fluorenylidene molecules in single crystals. J. Chem. Phys. 43:2006-16.
1969 With L. E. Closs. Induced dynamic nuclear spin polarization in reactions of photochemically and thermally generated triplet diphenylmethylene. J. Am. Chem. Soc. 91:4549-50.
With L. E. Closs. Induced dynamic nuclear spin polarization in photoreductions of benzophenone by toluene and ethylbenzene. J. Am. Chem. Soc. 91:4550-52.
A mechanism explaining nuclear spin polarizations in radical combination reactions. J. Am. Chem. Soc. 91:4552-54.
With A. D. Trifunac. Chemically induced nuclear spin polarization as a tool for determination of spin multiplicities of radical-pair precursors. J. Am. Chem. Soc. 91:4554-55.
1972 With C. E. Doubleday. Chemically induced dynamic nuclear spin polarization derived from biradicals generated by photochemical cleavage of cyclic ketones, and the observation of a solvent effect on signal intensities. J. Am. Chem. Soc. 94:9248-49.
1973 With C. E. Doubleday. Determination of the average singlet-triplet splitting in biradicals by measurement of the magnetic field dependence of CIDNP. J. Am. Chem. Soc. 95:2735-36.
1975 With S. G. Boxer. Nuclear magnetic resonance of photoexcited triplet states. I. The measurement of the rate of degenerate singlet-triplet exchange for anthracene in solution. J. Am. Chem. Soc. 97:3268-70.
1976 With B. E. Rabinow. Kinetic studies on diarylcarbenes. J. Am. Chem. Soc. 98:8190-98.
1979 With S. L. Buchwalter. Electron spin resonance and CIDNP studies on 1,3-cyclopentadiyls. A localized 1,3 carbon biradical system with a triplet ground state. Tunneling in carbon-carbon bond formation. J. Am. Chem. Soc. 101:4688-94.
1981 With E. V. Sitzmann. Measurements of degenerate radical ion-neutral molecule electron exchange by microsecond time-resolved CIDNP. Determination of relative hyperfine coupling constants of radical cations of chlorophylls and derivatives. J. Am. Chem. Soc. 103:3217-19.
With R. J. Miller. Laser flash photolysis with NMR detection. Submicrosecond time-resolved CIDNP: Kinetics of triplet states and biradicals. J. Am. Chem. Soc. 103:3586-88.
1983 With L. T. Calcaterra and J. R. Miller. Fast intramolecular electron
transfer in radical ions over long distances across rigid saturated hydrocarbon spacers. J. Am. Chem. Soc. 105:670-71.
1984 With J. R. Miller and L. T. Calcaterra. Intramolecular long-distance electron transfer in radical anions. The effects of free energy and solvent on the reaction rates. J. Am. Chem. Soc. 106:3047-49.
1986 With L. T. Calcaterra, N. J. Green, K. W. Penfield, and J. R. Miller. Distance, stereoelectronic effects, and the Marcus inverted region in intramolecular electron transfer in organic radical anions. J. Phys. Chem. 90:3673-83.
1988 With J. R. Miller. Intramolecular long-distance electron transfer in organic molecules. Science 240:440-47.
1989 With M. D. Johnson, J. R. Miller, and N. S. Green. Distance dependence of intramolecular hole and electron transfer in organic radical ions. J. Phys. Chem. 93:1173-76.
With M. D. Johnson, J. R. Miller, and P. Piotrowiak. A connection between intramolecular long-range electron, hole, and triplet energy transfers. J. Am. Chem. Soc. 111:3751-53.
With N. Liang and J. R. Miller. Correlating temperature dependence to free energy dependence of intramolecular long-range electron transfers. J. Am. Chem. Soc. 111:8740-41.
1990 With N. Liang and J. R. Miller. Temperature-independent long-range electron transfer reactions in the Marcus inverted region. J. Am. Chem. Soc. 112:5353-54.
1992 With M. D. E. Forbes and P. Piotrowiak. Spin and reaction dynamics in flexible polymethylene biradicals as studied by EPR, NMR, and optical spectroscopy and magnetic field effects. Measurements and mechanisms of scalar electron spin-spin coupling. J. Am. Chem. Soc. 114:3285-94.































