
4
Computational Tools
As a factual science, biological research involves the collection and analysis of data from potentially billions of members of millions of species, not to mention many trillions of base pairs across different species. As data storage and analysis devices, computers are admirably suited to the task of supporting this enterprise. Also, as algorithms for analyzing biological data have become more sophisticated and the capabilities of electronic computers have advanced, new kinds of inquiries and analyses have become possible.
4.1 THE ROLE OF COMPUTATIONAL TOOLS
Today, biology (and related fields such as medicine and pharmaceutics) are increasingly data-intensive—a trend that arguably began in the early 1960s.1 To manage these large amounts of data, and to derive insight into biological phenomena, biological scientists have turned to a variety of computational tools.
As a rule, tools can be characterized as devices that help scientists do what they know they must do. That is, the problems that tools help solve are problems that are known by, and familiar to, the scientists involved. Further, such problems are concrete and well formulated. As a rule, it is critical that computational tools for biology be developed in collaboration with biologists who have deep insights into the problem being addressed.
The discussion below focuses on three generic types of computational tools: (1) databases and data management tools to integrate large amounts of heterogeneous biological data, (2) presentation tools that help users comprehend large datasets, and (3) algorithms to extract meaning and useful information from large amounts of data (i.e., to find meaningful a signal in data that may look like noise at first glance). (Box 4.1 presents a complementary view of advances in computer sciences needed for next-generation tools for computational biology.)
|
1 |
The discussion in Section 4.1 is derived in part from T. Lenoir, “Shaping Biomedicine as an Information Science,” Proceedings of the 1998 Conference on the History and Heritage of Science Information Systems, M.E. Bowden, T.B. Hahn, and R.V. Williams, eds., ASIS Monograph Series, Information Today, Inc., Medford, NJ, 1999, pp. 27-45. |
|
Box 4.1 Data Representation
Analysis Tools
Visualization
Standards
Databases
SOURCE: U.S. Department of Energy, Report on the Computer Science Workshop for the Genomes to Life Program, Gaithersburg, MD, March 6-7, 2002; available at http://DOEGenomesToLife.org/compbio/. |
These examples are drawn largely from the area of cell biology. The reason is not that these are the only good examples of computational tools, but rather that a great deal of the activity in the field has been the direct result of trying to make sense out of the genomic sequences that have been collected to date. As noted in Chapter 2, the Human Genome Project—completed in draft in 2000—is arguably the first large-scale project of 21st century biology in which the need for powerful information technology was manifestly obvious. Since then, computational tools for the analysis of genomic data, and by extension data associated with the cell, have proliferated wildly; thus, a large number of examples are available from this domain.
4.2 TOOLS FOR DATA INTEGRATION2
As noted in Chapter 3, data integration is perhaps the most critical problem facing researchers as they approach biology in the 21st century.
|
2 |
Sections 4.2.1, 4.2.4, 4.2.6, and 4.2.8 embed excerpts from S.Y. Chung and J.C. Wooley, “Challenges Faced in the Integration of Biological Information,” in Bioinformatics: Managing Scientific Data, Z. Lacroix and T. Critchlow, eds., Morgan Kaufmann, San Francisco, CA, 2003. (Hereafter cited as Chung and Wooley, 2003.) |
4.2.1 Desiderata
If researcher A wants to use a database kept and maintained by researcher B, the “quick and dirty” solution is for researcher A to write a program that will translate data from one format into another. For example, many laboratories have used programs written in Perl to read, parse, extract, and transform data from one form into another for particular applications.3 Depending on the nature of the data involved and the structure of the source databases, writing such a program may require intensive coding.
Although such a fix is expedient, it is not scalable. That is, point-to-point solutions are not sustainable in a large community in which it is assumed that everyone wants to share data with everyone else. More formally, if there are N data sources to be integrated, and point-to-point solutions must be developed, N (N − 1)/2 translation programs must be written. If one data source changes (as is highly likely), N − 1 programs must be updated.
A more desirable approach to data integration is scalable. That is, a change in one database should not necessitate a change on the part of every research group that wants to use those data. A number of approaches are discussed below, but in general, Chung and Wooley argue that robust data integration systems must be able to
-
Access and retrieve relevant data from a broad range of disparate data sources;
-
Transform the retrieved data into a common data model for data integration;
-
Provide a rich common data model for abstracting retrieved data and presenting integrated data objects to the end-user applications;
-
Provide a high-level expressive language to compose complex queries across multiple data sources and to facilitate data manipulation, transformation, and integration tasks; and
-
Manage query optimization and other complex issues.
Sections 4.2.2, 4.2.4, 4.2.5, 4.2.6, and 4.2.8 address a number of different approaches to dealing with the data integration problem. These approaches are not, in general, mutually exclusive, and they may be usable in combination to improve the effectiveness of a data integration solution.
Finally, biological databases are always changing, so integration is necessarily an ongoing task. Not only are new data being integrated within the existing database structure (a structure established on the basis of an existing intellectual paradigm), but biology is a field that changes quickly—thus requiring structural changes in the databases that store data. In other words, biology does not have some “classical core framework” that is reliably constant. Thus, biological paradigms must be redesigned from time to time (on the scale of every decade or so) to keep up with advances, which means that no “gold standards” to organize data are built into biology. Furthermore, as biology expands its attention to encompass complexes of entities and events as well as individual entities and events, more coherent approaches to describing new phenomena will become necessary—approaches that bring some commonality and consistency to data representations of different biological entities—so that relationships between different phenomena can be elucidated.
As one example, consider the potential impact of “-omic” biology, biology that is characterized by a search for data completeness—the complete sequence of the human genome, a complete catalog of proteins in the human body, the sequencing of all genomes in a given ecosystem, and so on. The possibility of such completeness is unprecedented in the history of the life sciences and will almost certainly require substantial revisions to the relevant intellectual frameworks.
4.2.2 Data Standards
One obvious approach to data integration relies on technical standards that define representations of data and hence provide an understanding of data that is common to all database developers. For obvious reasons, standards are most relevant to future datasets. Legacy databases, which have been built around unique data definitions, are much less amenable to a standards-driven approach to data integration.
Standards are indeed an essential element of efforts to achieve data integration of future datasets, but the adoption of standards is a nontrivial task. For example, community-wide standards for data relevant to a certain subject almost certainly differ from those that might be adopted by individual laboratories, which are the focus of the “small-instrument, multi-data-source” science that characterizes most public-sector biological research.
Ideally, source data from these projects flow together into larger national or international data resources that are accessible to the community. Adopting community standards, however, entails local compromises (e.g., nonoptimal data structuring and semantics, greater expense), and the budgets that characterize small-instrument, single-data-source science generally do not provide adequate support for local data management and usually no support at all for contributions to a national data repository.
If data from such diverse sources are to be maintained centrally, researchers and laboratories must have incentives and support to adopt broader standards in the name of the community’s greater good. In this regard, funding agencies and journals have considerable leverage and through techniques such as requiring researchers to deposit data in conformance to community standards may be able to provide such incentives.
At the same time, data standards cannot resolve the integration problem by themselves even for future datasets. One reason is that in some fast-moving and rapidly changing areas of science (such as biology), it is likely that the data standards existing at any given moment will not cover some new dimension of data. A novel experiment may make measurements that existing data standards did not anticipate. (For example, sequence databases—by definition—do not integrate methylation data; and yet methylation is an essential characteristic of DNA that falls outside primary sequence information.) As knowledge and understanding advance, the meaning attached to a term may change over time. A second reason is that standards are difficult to impose on legacy systems, because legacy datasets are usually very difficult to convert to a new data standard and conversion almost always entails some loss of information.
As a result, data standards themselves must evolve as the science they support changes. Because standards cannot be propagated instantly throughout the relevant biological community, database A may be based on Version 12.1 of a standard, and database B on Version 12.4 of the “same” standard. It would be desirable if the differences between Versions 12.1 and 12.4 were not large and a basic level of integration could still be maintained, but this is not ensured in an environment in which options vary within standards, different releases and versions of products, and so on. In short, much of the devil of ensuring data integration is in the detail of implementation.
Experience in the database world suggests that standards gaining widespread acceptance in the commercial marketplace tend to have a long life span, because the marketplace tends to weed out weak standards before they become widely accepted. Once a standard is widely used, industry is often motivated to maintain compliance with this accepted standard, but standards created by niche players in the market tend not to survive. This point is of particular relevance in a fragmented research environment and suggests that standards established by strong consortia of multiple players are more likely to endure.
4.2.3 Data Normalization4
An important issue related to data standards is data normalization. Data normalization is the process through which data taken on the “same” biological phenomenon by different instruments, procedures, or researchers can be rendered comparable. Such problems can arise in many different contexts:
|
4 |
Section 4.2.3 is based largely on a presentation by C. Ball, “The Normalization of Microarray Data,” presented at the AAAS 2003 meeting in Denver, Colorado. |
-
Microarray data related to a given cell may be taken by multiple investigators in different laboratories.
-
Ecological data (e.g., temperature, reflectivity) in a given ecosystem may be taken by different instruments looking at the system.
-
Neurological data (e.g., timing and amplitudes of various pulse trains) related to a specific cognitive phenomenon may be taken on different individuals in different laboratories.
The simplest example of the normalization problem is when different instruments are calibrated differently (e.g., a scale in George’s laboratory may not have been zeroed properly, rendering mass measurements from George’s laboratory noncomparable to those from Mary’s laboratory). If a large number of readings have been taken with George’s scale, one possible fix (i.e., one possible normalization) is to determine the extent of the zeroing required and to add or subtract that correction to the already existing data. Of course, this particular procedure assumes that the necessary zeroing was constant for each of George’s measurements. The procedure is not valid if the zeroing knob was jiggled accidentally after half of the measurements had been taken.
Such biases in the data are systematic. In principle, the steps necessary to deal with systematic bias are straightforward. The researcher must avoid it as much as possible. Because complete avoidance is not possible, the researcher must recognize it when it occurs and then take steps to correct for it. Correcting for bias entails determining the magnitude and effect of the bias on data that have been taken and identifying the source of the bias so that the data already taken can be modified and corrected appropriately. In some cases, the bias may be uncorrectable, and the data must be discarded.
However, in practice, dealing with systematic bias is not nearly so straightforward. Ball notes that in the real world, the process goes something like this:
-
Notice something odd with data.
-
Try a few methods to determine magnitude.
-
Think of many possible sources of bias.
-
Wonder what in the world to do next.
There are many sources of systematic bias, and they differ depending on the nature of the data involved. They may include effects due to instrumentation, sample (e.g., sample preparation, sample choice), or environment (e.g., ambient vibration, current leakage, temperature). Section 3.3 describes a number of the systematic biases possible in microarray data, as do several references provided by Ball.5
There are many ways to correct for systematic bias, depending on the type of data being corrected. In the case of microarray studies, these ways include use of dye swap strategies, replicates and reference samples, experimental controls, consistent techniques, and sensible array and experiment design. Yet all
|
5 |
Ball’s AAAS presentation includes the following sources: T.B. Kepler, L. Crosby, and K.T. Morgan, “Normalization and Analysis of DNA Microarray Data by Self-consistency and Local Regression,” Genome Biololgy 3(7), RESEARCH0037.1- RESEARCH0037.12, 2002. Available at http://genomebiology.org/2002/3/7/research/0037.1; R. Hoffmann, T. Seidl, M. Dugas. “Profound Effect of Normalization on Detection of Differentially Expressed Genes in Oligonucleotide Microarray Data Analysis,” Genome Biolology 3(7):RESEARCH0033.1-RESEARCH0033.1-11. Available at http://genomebiology.com/2002/3/7/research/0033; C. Colantuoni, G. Henry, S. Zeger, and J. Pevsner, “Local Mean Normalization of Microarray Element Signal Intensities Across an Array Surface: Quality Control and Correction of Spatially Systematic Artifacts,” Biotechniques 32(6):1316-1320, 2002; B.P. Durbin, J.S. Hardin, D.M. Hawkins, and D.M. Rocke, “A Variance-Stabilizing Transformation for Gene-Expression Microarray Data,” Bioinformatics 18 (Suppl. 1):S105-S110, 2002; P.H. Tran, D.A. Peiffer, Y. Shin, L.M. Meek, J.P. Brody, and K.W. Cho, “Microarray Optimizations: Increasing Spot Accuracy and Automated Identification of True Microarray Signals,” Nucleic Acids Research 30(12):e54, 2002, available at http://nar.oupjournals.org/cgi/content/full/30/12/e54; M. Bilban, L.K. Buehler, S. Head, G. Desoye, and V. Quaranta, “Normalizing DNA Microarray Data,” Current Issues in Molecular Biology 4(2):57-64, 2002; J. Quackenbush, “Microarray Data Normalization and Transformation,” Nature Genetics Supplement 32:496-501, 2002. |
of these approaches are labor-intensive, and an outstanding challenge in the area of data normalization is to develop approaches to minimize systematic bias that demand less labor and expense.
4.2.4 Data Warehousing
Data warehousing is a centralized approach to data integration. The maintainer of the data warehouse obtains data from other sources and converts them into a common format, with a global data schema and indexing system for integration and navigation. Such systems have a long track record of success in the commercial world, especially for resource management functions (e.g., payroll, inventory). These systems are most successful when the underlying databases can be maintained in a controlled environment that allows them to be reasonably stable and structured. Data warehousing is dominated by relational database management systems (RDBMS), which offer a mature and widely accepted database technology and a standard high-level standard query language (SQL).
However, biological data are often qualitatively different from the data contained in commercial databases. Furthermore, biological data sources are much more dynamic and unpredictable, and few public biological data sources use structured database management systems. Data warehouses are often troubled by a lack of synchronization between the data they hold and the original database from which those data derive because of the time lag involved in refreshing the data warehouse store. Data warehousing efforts are further complicated by the issue of updates. Stein writes:6
One of the most ambitious attempts at the warehouse approach [to database integration] was the Integrated Genome Database (IGD) project, which aimed to combine human sequencing data with the multiple genetic and physical maps that were the main reagent for human genomics at the time. At its peak, IGD integrated more than a dozen source databases, including GenBank, the Genome Database (GDB) and the databases of many human genetic-mapping projects. The integrated database was distributed to end-users complete with a graphical front end…. The IGD project survived for slightly longer than a year before collapsing. The main reason for its collapse, as described by the principal investigator on the project (O. Ritter, personal communication, as relayed to Stein), was the database churn issue. On average, each of the source databases changed its data model twice a year. This meant that the IGD data import system broke down every two weeks and the dumping and transformation programs had to be rewritten—a task that eventually became unmanageable.
Also, because of the breadth and volume of biological databases, the effort involved in maintaining a comprehensive data warehouse is enormous—and likely prohibitive. Such an effort would have to integrate diverse biological information, such as sequence and structure, up to the various functions of biochemical pathways and genetic polymorphisms.
Still, data warehousing is a useful approach for specific applications that are worth the expense of intense data cleansing to remove potential errors, duplications, and semantic inconsistency.7 Two current examples of data warehousing are GenBank and the International Consortium for Brain Mapping (ICBM) (the latter is described in Box 4.2).
4.2.5 Data Federation
The data federation approach to integration is not centralized and does not call for a “master” database. Data federation calls for scientists to maintain their own specialized databases encapsulating their particular areas of expertise and retain control of the primary data, while still making it available to other researchers. In other words, the underlying data sources are autonomous. Data federation often
|
Box 4.2 In the human population, the brain varies structurally and functionally in currently undefined ways. It is clear that the size, shape, symmetry, folding pattern, and structural relationships of the systems in the human brain vary from individual to individual. This has been a source of considerable consternation and difficulty in research and clinical evaluations of the human brain from both the structural and the functional perspective. Current atlases of the human brain do not address this problem. Cytoarchitectural and clinical atlases typically use a single brain or even a single hemisphere as the reference specimen or target brain to which other brains are matched, typically with simple linear stretching and compressing strategies. In 1992, John Mazziotta and Arthur Toga proposed the concept of developing a probabilistic atlas from a large number of normal subjects between the ages of 18 and 90. This data acquisition has now been completed, and the value of such an atlas is being realized for both research and clinical purposes. The mathematical and software machinery required to develop this atlas of normal subjects is now also being applied to patient populations including individuals with Alzheimer’s disease, schizophrenia, autism, multiple sclerosis, and others. Talairach Atlas To date, more than 7,000 normal subjects have been entered into the Talairach atlas project and a wide range of datasets. These datasets contain detailed demographic histories of the subjects, results of general medical and neurological examinations, neuropsychiatric and neuropsychological evaluations, quantitative “handedness measurements”, and imaging studies. The imaging studies include multispectra 1 mm3 voxel-size magnetic resonance imaging (MRI) evaluations of the entire brain (T1, T2, and proton density pulse sequences). A subset of individuals also undergo functional MRI, cerebral blood flow position emission tomography (PET) and electroencephalogram (EEG) examinations (evoked potentials). Of these subjects, 5,800 individuals have also had their DNA collected and stored for future genotyping. As such, this database represents the most comprehensive evaluation of the structural and functional imaging phenotypes of the human brain in the normal population across a wide age span and very diverse social, economic, and racial groups. Participating laboratories are widely distributed geographically from Asia to Scandinavia, and include eight laboratories, in seven countries, on four continents. World Map of Sites A component of the World Map of Sites project involves the post mortem MRI imaging of individuals who have willed their bodies to science. Subsequent to MRI imaging, the brain is frozen and sectioned at a resolution of approximately 100 microns. Block face images are stored, and the sectioned tissue is stained for cytoarchitectural, chemoarchitectural, and differential myelin to produce microscopic maps of cellular anatomy, neuroreceptor or transmitter systems, and white matter tracts. These datasets are then incorporated into a target brain to which the in vivo brain studies are warped in three dimensions and labeled automatically. The 7,000 datasets are then placed in the standardized space, and probabilistic estimates of structural boundaries, volumes, symmetries, and shapes are computed for the entire population or any subpopulation (e.g., age, gender, race). In the current phase of the program, information is being added about in vivo chemoarchitecture (5-HT2A [5-hydroxytryptamine-2A] in vivo PET receptor imaging), in vivo white matter tracts (MRI-diffusion tensor imaging), vascular anatomy (magnetic resonance angiography and venography), and cerebral connections (transcranial magnetic stimulation-PET cerebral blood flow measurements). Target Brain The availability of 342 twin pairs in the dataset (half monozygotic and half dizygotic) along with DNA for genotyping provides the opportunity to understand structure-function relationships related to genotype and, therefore, provides the first large-scale opportunity to relate phenotype-genotype in behavior across a wide range of individuals in the human population. |
|
The development of similar atlases to evaluate patients with well-defined disease states allows the opportunity to compare the normal brain with brains of patients having cerebral pathological conditions, thereby potentially leading to enhanced clinical trials, automated diagnoses, and other clinical applications. Such examples have already emerged in patients with multiple sclerosis and epilepsy. An example in Alzheimer’s disease relates to a current hotly contested research question. Individuals with Alzheimer’s disease have a greater likelihood of having the genotype ApoE 4 (as opposed to ApoE 2 or 3). Having this genotype, however, is neither sufficient nor required for the development of Alzheimer’s disease. Individuals with Alzheimer’s disease also have small hippocampi, presumably because of atrophy of this structure as the disease progresses. The question of interest is whether individuals with the high-risk genotype (ApoE 4) have small hippocampi to begin with. This would be a very difficult hypothesis to test without the dataset described above. With the ICBM database, it is possible to study individuals from, for example, ages 20 to 40 and identify those with the smallest (lowest 5 percent) and largest (highest 5 percent) hippocampal volumes. This relatively small number of subjects could then be genotyped for ApoE alleles. If individuals with small hippocampi all had the genotype ApoE 4 and those with large hippocampi all had the genotype ApoE 2 or 3, this would be strong support for the hypothesis that individuals with the high-risk genotype for the development of Alzheimer’s disease have small hippocampi based on genetic criteria as a prelude to the development of Alzheimer’s disease. Similar genotype-imaging phenotype evaluations could be undertaken across a wide range of human conditions, genotypes, and brain structures. SOURCE: Modified from John C. Mazziotta and Arthur W. Toga, Department of Neurology, David Geffen School of Medicine, University of California, Los Angeles, personal communication to John Wooley, February 22, 2004. |
calls for the use of object-oriented concepts to develop data definitions, encapsulating the internal details of the data associated with the heterogeneity of the underlying data sources.8 A change in the representation or definition of the data then has minimal impact on the applications that access those data.
An example of a data federation environment is BioMOBY, which is based on two ideas.9 The first is the notion that databases provide bioinformatics services that can be defined by their inputs and outputs. (For example, BLAST is a service provided by GenBank that can be defined by its input—that is, an uncharacterized sequence—and by its output, namely, described gene sequences deposited in GenBank.) The second idea is that all database services would be linked to a central registry (MOBY Central) of services that users (or their applications) would query. From MOBY Central, a user could move from one set of input-output services to the next—for example, moving from one database that, given a sequence (the input), postulates the identity of a gene (the output), and from there to a database that, given a gene (the input), will find the same gene in multiple organisms (the output), and so on, picking up information as it moves through database services. There are limitations to the BioMOBY system’s ability to discriminate database services based the descriptions of inputs and outputs, and MOBY Central must be up and running 24 hours a day.10
4.2.6 Data Mediators/Middleware
In the middleware approach, an intermediate processing layer (a “mediator”) decouples the underlying heterogeneous, distributed data sources and the client layer of end users and applications.11 The mediator layer (i.e., the middleware) performs the core functions of data transformation and integration, and communicates with the database “wrappers” and the user application layer. (A “wrapper” is a software component associated with an underlying data source that is generally used to handle the tasks of access to specified data sources, extraction and retrieval of selected data, and translation of source data formats into a common data model designed for the integration system.)
The common model for data derived from the underlying data sources is the responsibility of the mediator. This model must be sufficiently rich to accommodate various data formats of existing biological data sources, which may include unstructured text files, semistructured XML and HTML files, and structured relational, object-oriented, and nested complex data models. In addition, the internal data model must facilitate the structuring of integrated biological objects to present to the user application layer. Finally, the mediator also provides services such as filtering, managing metadata, and resolving semantic inconsistency in source databases.
There are many flavors of mediator approaches in life science domains. IBM’s DiscoveryLink for the life sciences is one of the best known.12 The Kleisli system provides an internal nested complex data model and a high-power query and transformation language for data integration.13 K2 shares many design principles with Kleisli in supporting a complex data model, but adopts more object-oriented features.14 OPM supports a rich object model and a global schema for data integration.15 TAMBIS provides a global ontology (see Section 4.2.8 on ontologies) to facilitate queries across multiple data sources.16 TSIMMIS is a mediation system for information integration with its own data model (Object-Exchange Model, OEM) and query language.17
4.2.7 Databases as Models
A natural progression for databases established to meet the needs and interests of specialized communities, such as research on cell signaling pathways or programmed cell death, is the evolution of
|
11 |
G. Wiederhold, “Mediators in the Architecture of Future Information Systems,” IEEE Computer 25(3):38-49, 1992; G. Wiederhold and M. Genesereth, “The Conceptual Basis for Mediation Services,” IEEE Expert, Intelligent Systems and Their Applications 12(5):38-47, 1997. (Both cited in Chung and Wooley, 2003.) |
|
12 |
L.M. Haas et al., “DiscoveryLink: A System for Integrated access to Life Sciences Data Sources,” IBM Systems Journal 40(2):489-511, 2001. |
|
13 |
S. Davidson, C. Overton, V. Tannen, and L. Wong, “BioKleisli: A Digital Library for Biomedical Researchers,” International Journal of Digital Libraries 1(1):36-53, 1997; L. Wong, “Kleisli, a Functional Query System,” Journal of Functional Programming 10(1):19-56, 2000. (Both cited in Chung and Wooley, 2003.) |
|
14 |
J. Crabtree, S. Harker, and V. Tannen, “The Information Integration System K2,” available at http://db.cis.upenn.edu/K2/K2.doc; S.B. Davidson, J. Crabtree, B.P. Brunk, J. Schug, V. Tannen, G.C. Overton, and C.J. Stoeckert, Jr., “K2/Kleisli and GUS: Experiments in Integrated Access to Genomic Data Sources,” IBM Systems Journal 40(2):489-511, 2001. (Both cited in Chung and Wooley, 2003.) |
|
15 |
I-M.A. Chen and V.M. Markowitz, “An Overview of the Object-Protocol Model (OPM) and OPM Data Management Tools,” Information Systems 20(5):393-418, 1995; I-M.A. Chen, A.S. Kosky, V.M. Markowitz, and E. Szeto, “Constructing and Maintaining Scientific Database Views in the Framework of the Object-Protocol Model,” Proceedings of the Ninth International Conference on Scientific and Statistical Database Management, Institute of Electrical and Electronic Engineers, Inc., New York, 1997, pp. 237–248. (Cited in Chung and Wooley, 2003.) |
|
16 |
N.W. Paton, R. Stevens, P. Baker, C.A. Goble, S. Bechhofer, and A. Brass, “Query Processing in the TAMBIS Bioinformatics Source Integration System,” Proceedings of the 11th International Conference on Scientific and Statistical Database Management, IEEE, New York 1999, pp. 138-147; R. Stevens, P. Baker, S. Bechhofer, G. Ng, A. Jacoby, N.W. Paton, C.A. Goble, and A. Brass, “TAMBIS: Transparent Access to Multiple Bioinformatics Information Sources,” Bioinformatics 16(2):184-186, 2000. (Both cited in Chung and Wooley, 2003.) |
|
17 |
Y. Papakonstantinou, H. Garcia-Molina, and J. Widom, “Object Exchange Across Heterogeneous Information Sources,” Proceedings of the IEEE Conference on Data Engineering, IEEE, New York, 1995, pp. 251-260. (Cited in Chung and Wooley, 2003.) |
databases into models of biological activity. As databases become increasingly annotated with functional and other information, they lay the groundwork for model formation.
In the future, such “database models” are envisioned as the basis of informed predictions and decision making in biomedicine. For example, physicians of the future may use biological information systems (BISs) that apply known interactions and causal relationships among proteins that regulate cell division to changes in an individual’s DNA sequence, gene expression, and proteins in an individual tumor.18 The physician might use this information together with the BIS to support a decision on whether the inhibition of a particular protein kinase is likely to be useful for treating that particular tumor.
Indeed, a major goal in the for-profit sector is to create richly annotated databases that can serve as testbeds for modeling pharmaceutical applications. For example, Entelos has developed PhysioLab, a computer model system consisting of a large set (more than 1,000) of ordinary nonlinear differential equations.19 The model is a functional representation of human pathophysiology based on current genomic, proteomic, in vitro, in vivo, and ex vivo data, built using a top-down, disease-specific systems approach that relates clinical outcomes to human biology and physiology. Starting with major organ systems, virtual patients are explicit mathematical representations of a particular phenotype, based on known or hypothesized factors (genetic, life-style, environmental). Each model simulates up to 60 separate responses previously demonstrated in human clinical studies.
In the neuroscience field, Bower and colleagues have developed the Modeler’s Workspace,20 which is based on a notion that electronic databases must provide enhanced functionality over traditional means of distributing information if they are to be fully successful. In particular, Bower et al. believe that computational models are an inherently more powerful medium for the electronic storage and retrieval of information than are traditional online databases.
The Modeler’s Workspace is thus designed to enable researchers to search multiple remote databases for model components based on various criteria; visualize the characteristics of the components retrieved; create new components, either from scratch or derived from existing models; combine components into new models; link models to experimental data as well as online publications; and interact with simulation packages such as GENESIS to simulate the new constructs.
The tools contained in the Workspace enable researchers to work with structurally realistic biological models, that is, models that seek to capture what is known about the anatomical structure and physiological characteristics of a neural system of interest. Because they are faithful to biological anatomy and physiology, structurally realistic models are a means of storing anatomical and physiological experimental information.
For example, to model a part of the brain, this modeling approach starts with a detailed description of the relevant neuroanatomy, such as a description of the three-dimensional structure of the neuron and its dendritic tree. At the single-cell level, the model represents information about neuronal morphology, including such parameters as soma size, length of interbranch segments, diameter of branches, bifurcation probabilities, and density and size of dendritic spines. At the neuronal network level, the model represents the cell types found in the network and the connectivity among them. The model must also incorporate information regarding the basic physiological behavior of the modeled structure—for example, by tuning the model to replicate neuronal responses to experimentally derived data.
With such a framework in place, a structural model organizes data in ways that make manifestly obvious how those data are related to neural function. By contrast, for many other kinds of databases it is not at all obvious how the data contained therein contribute to an understanding of function. Bower
|
18 |
R. Brent and D. Endy, “Modelling Cellular Behaviour,” Nature 409:391-395, 2001. |
|
19 |
See, for example, http://www.entelos.com/science/physiolabtech.html. |
|
20 |
M. Hucka, K. Shankar, D. Beeman, and J.M. Bower, “The Modeler’s Workspace: Making Model-Based Studies of the Nervous System More Accessible,” Computational Neuroanatomy: Principles and Methods, G.A. Ascoli, ed., Humana Press, Totowa, NJ, 2002, pp. 83-103. |
and colleagues argue that “as models become more sophisticated, so does the representation of the data. As models become more capable, they extend our ability to explore the functional significance of the structure and organization of biological systems.”21
4.2.8 Ontologies
Variations in language and terminology have always posed a great challenge to large-scale, comprehensive integration of biological findings. In part, this is due to the fact that scientists operate, with a data- and experience-driven intuition that outstrips the ability of language to describe. As early as 1952, this problem was recognized:
Geneticists, like all good scientists, proceed in the first instance intuitively and … their intuition has vastly outstripped the possibilities of expression in the ordinary usages of natural languages. They know what they mean, but the current linguistic apparatus makes it very difficult for them to say what they mean. This apparatus conceals the complexity of the intuitions. It is part of the business of genetical methodology first to discover what geneticists mean and then to devise the simplest method of saying what they mean. If the result proves to be more complex than one would expect from the current expositions, that is because these devices are succeeding in making apparent a real complexity in the subject matter which the natural language conceals.22
In addition, different biologists use language with different levels of precision for different purposes. For instance, the notion of “identity” is different depending on context.23 Two geneticists may look at a map of human chromosome 21. A year later, they both want to look at the same map again. But to one of them, “same” means exactly the same map (same data, bit for bit); to the other, it means the current map of the same biological object, even if all of the data in that map have changed. To a protein chemist, two molecules of beta-hemoglobin are the same because they are composed of exactly the same sequence of amino acids. To a biologist, the same two molecules might be considered different because one was isolated from a chimpanzee and the other from a human.
To deal with such context-sensitive problems, bioinformaticians have turned to ontologies. An ontology is a description of concepts and relationships that exist among the concepts for a particular domain of knowledge.24 Ontologies in the life sciences serve two equally important functions. First, they provide controlled, hierarchically structured vocabularies for terminology that can be used to describe biological objects. Second, they specify object classes, relations, and functions in ways that capture the main concepts of and relationships in a research area.
4.2.8.1 Ontologies for Common Terminology and Descriptions
To associate concepts with the individual names of objects in databases, an ontology tool might incorporate a terminology database that interprets queries and translates them into search terms consistent with each of the underlying sources. More recently, ontology-based designs have evolved from static dictionaries into dynamic systems that can be extended with new terms and concepts without modification to the underlying database.
|
21 |
M. Hucka, K. Shankar, D. Beeman, and J.M. Bower, “The Modeler’s Workspace,” 2002. |
|
22 |
J.H. Woodger, Biology and Language, Cambridge University Press, Cambridge, UK, 1952. |
|
23 |
R.J. Robbins, “Object Identity and Life Science Research,” position paper submitted for the Semantic Web for Life Sciences Workshop, October 27-28 2004, Cambridge, MA, available at http://lists.w3.org/Archives/Public/public-swls-ws/2004Sep/att-0050/position-01.pdf. |
|
24 |
The term “ontology” is a philosophical term referring to the subject of existence. The computer science community borrowed the term to refer to “specification of a conceptualization” for knowledge sharing in artificial intelligence. See, for example, T.R. Gruber, “A Translation Approach to Portable Ontology Specification,” Knowledge Acquisition 5(2):199-220, 1993. (Cited in Chung and Wooley, 2003.) |
A feature of ontologies that facilitates the integration of databases is the use of a hierarchical structure that is progressively specialized; that is, specific terms are defined as specialized forms of general terms. Two different databases might not extend their annotation of a biological object to the same level of specificity, but the databases can be integrated by finding the levels within the hierarchy that share a common term.
The naming dimension of ontologies has been common to research in the life sciences for much of its history, although the term itself has not been widely used. Chung and Wooley note the following, for example:
-
The Linnaean system for naming of species and organisms in taxonomy is one of the oldest ontologies.
-
The nomenclature committee for the International Union of Pure and Applied Chemistry (IUPAC) and the International Union of Biochemistry and Molecular Biology (IUBMB) make recommendations on organic, biochemical, and molecular biology nomenclature, symbols, and terminology.
-
The National Library of Medicine Medical Subject Headings (MeSH) provides the most comprehensive controlled vocabularies for biomedical literature and clinical records.
-
A division of the College of American Pathologists oversees the development and maintenance of a comprehensive and controlled terminology for medicine and clinical information known as SNOMED (Systematized Nomenclature of Medicine).
-
The Gene Ontology Consortium25 seeks to create an ontology to unify work across many genomic projects—to develop controlled vocabulary and relationships for gene sequences, anatomy, physical characteristics, and pathology across the mouse, yeast, and fly genomes.26 The consortium’s initial efforts focus on ontologies for molecular function, biological process, and cellular components of gene products across organisms and are intended to overcome the problems associated with inconsistent terminology and descriptions for the same biological phenomena and relationships.
Perhaps the most negative aspect of ontologies is that they are in essence standards, and hence take a long time to develop—and as the size of the relevant community affected by the ontology increases, so does development time. For example, the ecological and biodiversity communities have made substantial progress in metadata standards, common taxonomy, and structural vocabulary with the help of National Science Foundation and other government agencies.27 By contrast, the molecular biology community is much more diverse, and reaching a community-wide consensus has been much harder.
An alternative to seeking community-wide consensus is to seek consensus in smaller subcommunities associated with specific areas of research such as sequence analysis, gene expression, protein pathways, and so on.28 These efforts usually adopt a use-case and open-source approach for community input. The ontologies are not meant to be mandatory, but instead to serve as a reference framework from which further development can proceed.
|
25 |
See www.geneontology.org. |
|
26 |
M. Ashburner, C.A. Ball, J.A. Blacke, D. Botstein, H. Butler, J.M. Cherry, A.P. Davis, et al., “Gene Ontology: Tool for the Unification of Biology,” Nature Genetics 25(1):25–29, 2000. (Cited in Chung and Wooley, 2003.) |
|
27 |
J.L. Edwards, M.A. Lane, and E.S. Nielsen, “Interoperability of Biodiversity Databases: Biodiversity Information on Every Desk,” Science 289(5488):2312-2314, 2000; National Biological Information Infrastructure (NBII), available at http://www.nbii.gov/disciplines/systematics.html; Federal Geographic Data Committee (FGDC), available at http://www.fgdc.gov/. (All cited in Chung and Wooley, 2003.) |
|
28 |
Gene Expression Ontology Working Group, see http://www.mged.org/; P.D. Karp, M. Riley, S.M. Paley, and A. Pellegrini-Toole, “The MetaCyc Database,” Nucleic Acids Research 30(1):59-61, 2002; P.D. Karp, M. Riley, M. Saier, I.T. Paulsen, J. Collado-Vides, S.M. Paley, A. Pellegrini-Toole, et al., “The EcoCyc Database,” Nucleic Acids Research 30(1):56-58, 2002; D.E. Oliver, D.L. Rubin, J.M. Stuart, M. Hewett, T.E. Klein, and R.B. Altman, “Ontology Development for a Pharmacogenetics Knowledge Base,” Pacific Symposium on Biocomputing 65-76, 2002. (All cited in Chung and Wooley, 2003.) |
An ontology developed by one subcommunity inevitably leads to interactions with related ontologies and the need to integrate. For example, consider the concept of homology. In traditional evolutionary biology, “analogy” is used to describe things that are identical by function and “homology” is used to identify things that are identical by descent. However, in considering DNA, function and descent are both captured in the DNA sequence, and therefore to molecular biologists, homology has come to mean simply similarity in sequence, regardless of whether this is due to convergence or ancestry. Thus, the term “homologous” means different things in molecular biology and evolutionary biology.29 More broadly, a brain ontology will inevitably relate to ontologies of other anatomic structures or at the molecular level sharing ontologies for genes and proteins.30
Difficulties of integrating diverse but related databases thus are transformed into analogous difficulties in integrating diverse but related ontologies, but since each ontology represents the integration of multiple databases relevant to the field, the integration effort at the higher level is more encompassing. At the same time, it is also more difficult, because the implications of changes in fundamental concepts—which will be necessary in any integration effort—are much more far-reaching than analogous changes in a database. That is, design compromises in the development of individual ontologies might make it impossible to integrate the ontologies without changes to some of their basic components. This would require undoing the ontologies, then redoing them to support integration.
These points relate to semantic interoperability, which is an active area of research in computer science.31 Information integration across multiple biological disciplines and subdisciplines would depend on the close collaborations of domain experts and information technology professionals to develop algorithms and flexible approaches to bridge the gaps between multiple biological ontologies. In recent years, a number of life science researchers have come to believe in the potential of the Semantic Web for integrating biological ontologies, as described in Box 4.3.
A sample collection of ontology resources for controlled vocabulary purposes in the life sciences is listed in Table 4.1.
4.2.8.2 Ontologies for Automated Reasoning
Today, it is standard practice to store biological data in databases; no one would deny that the volume of available data is far beyond the capabilities of human memory or written text. However, even as the volume of analytic and theoretical results drawn from these data (such as inferred genetic regulatory, metabolic, and signaling network relationships) grows, it will become necessary to store such information as well in a format suitable for computational access.
The essential rationale underlying automated reasoning is that reasoning one’s way through all of the complexity inherent in biological organisms is very difficult, and indeed may be, for all practical purposes, impossible for the knowledge bases that are required to characterize even the simplest organisms. Consider, for example, the networks related to genetic regulation, metabolism, and signaling of an organism such as Escherichia coli. These networks are too large for humans to reason about in their totality, which means that it is increasingly difficult for scientists to be certain about global network properties. Is the model complete? Is it consistent? Does it explain all of the data? For example, the database of known molecular pathways in E. coli contains many hundreds of connections, far more than most researchers could remember, much less reason about.
|
Box 4.3 The Semantic Web seeks to create a universal medium for the exchange of machine-understandable data of all types, including biological data. Using Semantic Web technology, programs can share and process data even when they have been designed totally independently. The semantic web involves a Resource Description Framework (RDF), an RDF Schema language, and the Web Ontology language (OWL). RDF and OWL are Semantic Web standards that provide a framework for asset management, enterprise integration and the sharing and reuse of data on the Web. Furthermore, a standardized query language for RDF enables the “joining” of decentralized collections of RDF data. The underlying technology foundation of these languages is that of URLs, XML, and XML name spaces. Within the life sciences, the notion of a life sciences identifier (LSID) is intended to provide a straightforward approach to naming and identifying data resources stored in multiple, distributed data stores in a manner that overcomes the limitations of naming schemes in use today. LSIDs are persistent, location-independent, resource identifiers for uniquely naming biologically significant resources including but not limited to individual genes or proteins, or data objects that encode information about them. The life sciences pose a particular challenge for data integration because the semantics of biological knowledge are constantly changing. For example, it may be known that two proteins bind to each other. But this fact could be represented at the cellular level, the tissue level, and the molecular level depending on the context in which that fact was important. The Semantic Web is intended to allow for evolutionary change in the relevant ontologies as new science emerges without the need for consensus. For example, if Researcher A states (and encodes using Semantic Web technology) a relationship between a protein and a signaling cascade with which Researcher B disagrees, Researcher B can instruct his or her computer to ignore (perhaps temporarily) the relationship encoded by Researcher A in favor (perhaps) of a relationship that is defined only locally. An initiative coordinated by the World Wide Web Consortium seeks to explore how Semantic Web technologies can be used to reduce the barriers and costs associated with effective data integration, analysis, and collaboration in the life sciences research community, to enable disease understanding, and to accelerate the development of therapies. A meeting in October 2004 on the Semantic Web and the life sciences concluded that work was needed in two high-priority areas.
SOURCES: The Semantic Web Activity Statement, available at http://www.w3.org/2001/sw/Activity; Life Sciences Identifiers RFP Response, OMG Document lifesci/2003-12-02, January 12, 2004, available at http://www.omg.org/docs/lifesci/03-12-02.doc#_Toc61702471; John Wilbanks, Science Commons, Massachusetts Institute of Technology, personal communication, April 4, 2005. |
TABLE 4.1 Biological Ontology Resources
|
Organization |
Descriptions |
|
Human Genome Organization (HUGO) Gene Nomenclature Committee (HGNC): http://www.gene.ucl.ac.uk/nomenclature/ |
HGNC is responsible for the approval of a unique symbol for each gene and designate description of genes. Aliases for genes are also listed in the database. |
|
Gene Ontology Consortium (GO): http://www.geneontology.org |
The purpose of GO is to develop ontologies describing the molecular function, biological process, and cellular component of genes and gene products for eukaryotes. Members include genome databases of fly, yeast, mouse, worm, and Arabidopsis. |
|
Plant Ontology Consortium: http://www.plantontology.org |
This consortium will produce structured, controlled vocabularies applied to plant-based database information. |
|
Microarrey Gene Expression Data (MGED) Society Ontology Working Group: http://www.mged.org/ |
The MGED group facilitates the adoption of standards for DNA-microarray experiment annotation and data representation, as well as the introduction of standard expertmental controls and data normalization methods. |
|
NIBII (National Biological Information Infrastructure): http://www.nbii.gov/disciplines/systematics.html |
NBII provides links to taxonomy sites for all biological disciplines. |
|
ITIS (Integrated Taxonomic Information System): http://www.itis.usda.gov/ |
ITIS provides taxonomic information on plants, animals, and microbes of North America and the world. |
|
MeSH (Medical Subject Headings): http://www.nlm.nih.gov/mesh/meshhome.html |
MeSH is a controlled vocabulary established by the National Library of Medicine (NLM) and used for indexing articles, cataloging books and other holdings, and searching MeSH-indexed databases, including MEDLINE. |
|
SNOMED (Systematized Nomenclature of Medicine): http://www.snomed.org/ |
SNOMED is recognized globally as a comprehensive, multiaxial, controlled terminology created for the indexing of the entire medical record. |
|
International Classification of Diseases, Ninth Revision, Clinical Modification (ICD-9-CM): http://www.cdc.gov/nchs/about/otheract/lcd9/abtlcd9.htm |
ICD-9-CM is the official system of assigning codes to diagnoses and procedures associated with hospital utilization in the United States. It is published by the U.S. National Center for Health Statistics. |
|
International Union of Pure and Applied Chemistry (IUPAQ) |
IUPAC and IUBMB make recommendations on organic, biochemical, and molecular biology nomenclature, symbols, and terminology. |
|
International Union of Biochemistry and Molecular Biology (IUBMB) Nomenclature Committee: http://www.chem.q-mul.ac.uk/iubmb/ |
|
|
PharmGKB ( Pharmacogenetics Knowledge Base: http://pharmgkb.org/ |
PharmGKB, develops ontologies for pharmacogenetics and pharmacogenomics. |
|
Organization |
Descriptions |
|
mmCEF (Macromolecular Crystallographic Information File): http://pdb.rutgers.edu/mmcif/ http://www.iucr.ac.ukliucr-top/cif/index.html |
The information file mmCEF is sponsored by IUCr (International Union of Crystallography) to provide a dictionary for data items relevant to macromolecular crystallographic experiments. |
|
LocusLink: http://www.ncbi.nlm.nih.gov/LocusLink/ |
LocusLink contains gene-centered resources, including nomenclature and aliases for genes. |
|
Protégé-2000: http://protege.stanford.edu |
Protégé-2000 is a tool that allows the user to construct a domain ontology that can be extended to access embedded applications in other knowledge-based systems. A number of biomedical ontologies have been constructed with this system, but it can be applied to other domains as well. |
|
TAMBIS aims to aid researchers in the biological sciences by providing a single access point for biological information sources around the world. The access point will be a single Web-based interface that acts as a single information source. It will find appropriate sources of information for user queries and phrase the user questions for each source, returning the results in a consistent manner which will include details of the information source. |
By representing working hypotheses, derived results, and the evidence that supports and refutes them in machine-readable representations, researchers can uncover correlations in and make inferences about independently conducted investigations of complex biological systems that would otherwise remain undiscovered by relying simply on serendipity or their own reasoning and memory capacities.32 In principle, software can read and operate on these representations, determining properties in a way similar to human reasoning, but able to consider hundreds or thousands of elements simultaneously. Although automated reasoning can potentially predict the response of a biological system to a particular stimulus, it is particularly useful for discovering inconsistencies or missing relations in the data, establishing global properties of networks, discovering predictive relationships between elements, and inferring or calculating the consequences of given causal relationships.33 As the number of discovered pathways and molecular networks increases and the questions of interest to researchers become more about global properties of organisms, automated reasoning will become increasingly useful.
Symbolic representations of biological knowledge—ontologies—are a foundation for such efforts. Ontologies contain names and relationships of the many objects considered by a theory, such as genes, enzymes, proteins, transcription, and so forth. By storing such an ontology in a symbolic machine-
|
32 |
L. Hunter, “Ontologies for Programs, Not People,” Genome Biology 3(6):1002.1-1002.2, 2002. |
|
33 |
As shown in Chapter 5, simulations are also useful for predicting the response of a biological system to various stimuli. But simulations instantiate procedural knowledge (i.e., how to do something), whereas the automated reasoning systems discussed here operate on declarative knowledge (i.e., knowledge about something). Simulations are optimized to answer a set of questions that is narrower than those that can be answered by automated reasoning systems—namely, predictions about the subsequent response of a system to a given stimulus. Automated reasoning systems can also answer such questions (though more slowly), but in addition they can answer questions such as, What part of a network is responsible for this particular response?, presuming that such (declarative) knowledge is available in the database on which the systems operate. |
readable form and making use of databases of biological data and inferred networks, software based on artificial intelligence research can make complex inferences using these encoded relationships, for example, to consider statements written in that ontology for consistency or to predict new relationships between elements.34 Such new relationships might include new metabolic pathways, regulatory relationships between genes, signaling networks, or other relationships. Other approaches rely on logical frameworks more expressive than database queries and are able to reason about explanations for a given feature or suggest plans for intervention to reach a desired state.35
Developing an ontology for automated reasoning can make use of many different sources. For example, inference from gene-expression data using Bayesian networks can take advantage of online sources of information about the likely probabilistic dependencies among expression levels of various genes.36 Machine-readable knowledge bases can be built from textbooks, review articles, or even the Oxford Dictionary of Molecular Biology. The rapidly growing volume of publications in the biological literature is another important source, because inclusion of the knowledge in these publications helps to uncover relationships among various genes, proteins, and other biological entities referenced in the literature.
An example of ontologies for automated reasoning is the ontology underlying the EcoCyc database. The EcoCyc Pathway Database (http://ecocyc.org) describes the metabolic transport, and genetic regulatory networks of E. coli. EcoCyc structures a scientific theory about E. coli within a formal ontology so that the theory is available for computational analysis.37 Specifically, EcoCyc describes the genes and proteins of E. coli as well as its metabolic pathways, transport functions, and gene regulation. The underlying ontology encodes a diverse array of biochemical processes, including enzymatic reactions involving small molecule substrates and macromolecular substrates, signal transduction processes, transport events, and mechanisms of regulation of gene expression.38
4.2.9 Annotations and Metadata
Annotation is auxiliary information associated with primary information contained in a database. Consider, for example, the human genome database. The primary database consists of a sequence of some 3 billion nucleotides, which contains genes, regulatory elements, and other material whose function is unknown. To make sense of this enormous sequence, the identification of significant patterns within it is necessary. Various pieces of the genome must be identified, and a given sequence might be annotated as translation (e.g., “stop”), transcription (e.g., “exon” or “intron”), variation (“insertion”), structural (“clone”), similarity, repeat, or experimental (e.g., “knockout,” “transgenic”). Identifying a particular nucleotide sequence as a gene would itself be an annotation, and the protein corresponding to it, including its three-dimensional structure characterized as a set of coordinates of the protein’s atoms, would also be an annotation. In short, the sequence database includes the raw sequence data, and the annotated version adds pertinent information such as gene coded for, amino acid sequence, or other commentary to the database entry of raw sequence of DNA bases.39
|
34 |
P.D. Karp, “Pathway Databases: A Case Study in Computational Symbolic Theories,” Science 293(5537):2040-2044, 2001. |
|
35 |
C. Baral, K. Chancellor, N. Tran, N.L. Tran, A. Joy, and M. Berens, “A Knowledge Based Approach for Representing and Reasoning About Signaling Networks,” Bioinformatics 20(Suppl. 1):I15-I22, 2004. |
|
36 |
E. Segal, B. Taskar, A. Gasch, N. Friedman, and D. Koller, “Rich Probabilistic Models for Gene Expression,” Bioinformatics 17(Supp. 1):S243-S252, 2001. (Cited in Hunter, “Ontologies for Programs, Not People,” 2002, Footnote 32.) |
|
37 |
P.D. Karp, “Pathway Databases: A Case Study in Computational Symbolic Theories,” Science 293(5537):2040-2044, 2001; P.D. Karp, M. Riley, M. Saier, I.T. Paulsen, J. Collado-Vides, S.M. Paley, A Pellegrini-Toole, et al., “The EcoCyc Database,” Nucleic Acids Research 30(1):56-58, 2002. |
|
38 |
P.D. Karp, “An Ontology for Biological Function Based on Molecular Interactions,” Bioinformatics 16(3):269–285, 2000. |
|
39 |
See http://www.biochem.northwestern.edu/holmgren/Glossary/Definitions/Def-A/Annotation.html. |
Although the genomic research community uses annotation to refer to auxiliary information that has biological function or significance, annotation could also be used as a way to trace the provenance of data (discussed in greater detail in Section 3.7). For example, in a protein database, the utility of an entry describing the three-dimensional structure of a protein would be greatly enhanced if entries also included annotations that described the quality of data (e.g., their precision), uncertainties in the data, the physical and chemical properties of the protein, various kinds of functional information (e.g., what molecules bind to the protein, location of the active site), contextual information such as where in a cell the protein is found and in what concentration, and appropriate references to the literature.
In principle, annotations can often be captured as unstructured natural language text. But for maximum utility, machine-readable annotations are necessary. Thus, special attention must be paid to the design and creation of languages and formats that facilitate machine processing of annotations. To facilitate such processing, a variety of metadata tools are available. Metadata—or literally “data about data”—are anything that describes data elements or data collections, such as the labels of the fields, the units used, the time the data were collected, the size of the collection, and so forth. They are invaluable not only for increasing the life span of data (by making it easier or even possible to determine the meaning of a particular measurement), but also for making datasets comprehensible to computers. The National Biological Information Infrastructure (NBII)40 offers the following description:
Metadata records preserve the usefulness of data over time by detailing methods for data collection and data set creation. Metadata greatly minimize duplication of effort in the collection of expensive digital data and foster sharing of digital data resources. Metadata supports local data asset management such as local inventory and data catalogs, and external user communities such as Clearinghouses and websites. It provides adequate guidance for end-use application of data such as detailed lineage and context. Metadata makes it possible for data users to search, retrieve, and evaluate data set information from the NBII’s vast network of biological databases by providing standardized descriptions of geospatial and biological data.
A popular tool for the implementation of controlled metadata vocabularies is the extensible markup language (XML).41 XML offers a way to serve and describe data in a uniform and automatically parsable format and provides an open-source solution for moving data between programs. Although XML is a language for describing data, the descriptions of data are articulated in XML-based vocabularies.
Such vocabularies are useful for describing specific biological entities along with experimental information associated with those entities. Some of the vocabularies have been developed in association with specialized databases established by the community. Because of their common basis in XML, however, one vocabulary can be translated to another using various tools, for example, the XML style sheet language transformation, or XSLT.42
Examples of such XML-based dialects include the BIOpolymer Markup Language (BIOML),43 designed for annotating the sequences of biopolymers (e.g., genes, proteins), in such a way that all information about a biopolymer can be logically and meaningfully associated with it. Much like HTML, the language uses tags such as <protein>, <subunit>, and <peptide> to describe elements of a biopolymer along with a series of attributes.
The Microarray Markup Language (MAML) was created by a coalition of developers (www.beahmish.lbl.gov) to meet community needs for sharing and comparing the results of gene expression experiments. That community proposed the creation of a Microarray Gene Expression Database and defined the minimum information about a microarray experiment (MIAME) needed to enable
sharing. Consistent with the MIAME standards proposed by microarray users, MAML can be used to describe experiments and results from all types of DNA arrays.
The Systems Biology Markup Language, (SBML) is used to represent and model information in systems simulation software, so that models of biological systems can be exchanged by different software programs (e.g., E-Cell, StochSim). The SBML language, developed by the Caltech ERATO Kiranto systems biology Project,44 is organized around five categories of information: model, compartment, geometry, specie, and reaction.
A downside of XML is that only a few of the largest and most used databases (e.g., a GenBank) support an XML interface. Other databases whose existence predates XML keep most of their data in flat files. But this reality is changing, and database researchers are working to create conversion tools and new database platforms based on XML. Additional XML-based vocabularies and translation tools are needed.
The data annotation process is complex and cumbersome when large datasets are involved, and some efforts have been made to reduce the burden of annotation. For example, the Distributed Annotation System (DAS) is a Web service for exchanging genome annotation data from a number of distributed databases. The system depends on the existence of a “reference sequence” and gathers “layers” of annotation about the sequence that reside on third-party servers and are controlled by each annotation provider. The data exchange standard (the DAS XML specification) enables layers to be provided in real time from the third-party servers and overlaid to produce a single integrated view by a DAS client. Success in the effort depends on the willingness of investigators to contribute annotation information recorded on their respective servers, and on users’ learning about the existence of a DAS server (e.g., through ad hoc mechanisms such as link lists). DAS is also more or less specific to sequence annotation and is not easily extended to other biological objects.
Today, when biologists archive a newly discovered gene sequence in GenBank, for example, they have various types of annotation software at their disposal to link it with explanatory data. Next-generation annotation systems will have to do this for many other genome features, such as transcription-factor binding sites and single nucleotide polymorphisms (SNPs), that most of today’s systems don’t cover at all. Indeed, these systems will have to be able to create, annotate, and archive models of entire metabolic, signaling, and genetic pathways. Next-generation annotation systems will have to be built in a highly modular and open fashion, so that they can accommodate new capabilities and new data types without anyone’s having to rewrite the basic code.
4.2.10 A Case Study: The Cell Centered Database45
To illustrate the notions described above, it is helpful to consider an example of a database effort that implements many of them. Techniques such as electron tomography are generating large amounts of exquisitely detailed data on cells and their macromolecular organization that have to be exposed to the greater scientific community. However, very few structured data repositories for community use exist for the type of cellular and subcellular information produced using light and electron microscopy. The Cell Centered Database (CCDB) addresses this need by developing a database for three-dimensional light and electron microscopic information.46
|
44 |
|
|
45 |
Section 4.2.10 is adapted largely from M.E. Martone, S.T. Peltier, and M.H. Ellisman, “Building Grid Based Resources for Neurosciences,” National Center for Microscopy and Imaging Research, Department of Neurosciences, University of California, San Diego, unpublished and undated working paper. |
|
46 |
M.E. Martone, A. Gupta, M. Wong, X. Qian, G. Sosinsky, B. Ludascher, and M.H. Ellisman, “A Cell-Centered Database for Electron Tomographic Data,” Journal of Structural Biology 138(1-2):145-155, 2002; M.E. Martone, S. Zhang, S. Gupta, X. Qian, H. He, D.A. Price, M. Wong, et al., “The Cell Centered Database: A Database for Multiscale Structural and Protein Localization Data from Light and Electron Microscopy,” Neuroinformatics 1(4):379-396, 2003. |
The CCDB contains structural and protein distribution information derived from confocal, multiphoton, and electron microscopy, including correlated microscopy. Its main mission is to provide a means to make high-resolution data derived from electron tomography and high-resolution light microscopy available to the scientific community, situating itself between whole brain imaging databases such as the MAP project47 and protein structures determined from electron microscopy, nuclear magnetic resonance (NMR) spectroscopy, and X-ray crystallography (e.g., the Protein Data Bank and EMBL).
The CCDB serves as a research prototype for investigating new methods of representing imaging data in a relational database system so that powerful data-mining approaches can be employed for the content of imaging data. The CCDB data model addresses the practical problem of image management for the large amounts of imaging data and associated metadata generated in a modern microscopy laboratory. In addition, the data model has to ensure that data within the CCDB can be related to data taken at different scales and modalities.
The data model of the CCDB was designed around the process of three-dimensional reconstruction from two-dimensional micrographs, capturing key steps in the process from experiment to analysis. (Figure 4.1 illustrates the schema-entity relationship for the CCDB.) The types of imaging data stored in the CCDB are quite heterogeneous, ranging from large-scale maps of protein distributions taken by confocal microscopy to three-dimensional reconstruction of individual cells, subcellular structures, and organelles. The CCDB can accommodate data from tissues and cultured cells regardless of tissue of origin, but because of the emphasis on the nervous system, the data model contains several features specialized for neural data. For each dataset, the CCDB stores not only the original images and three-dimensional reconstruction, but also any analysis products derived from these data, including segmented objects and measurements of quantities such as surface area, volume, length, and diameter. Users have access to the full resolution imaging data for any type of data, (e.g., raw data, three-dimensional reconstruction, segmented volumes), available for a particular dataset.
For example, a three-dimensional reconstruction is viewed as one interpretation of a set of raw data that is highly dependent on the specimen preparation and imaging methods used to acquire it. Thus, a single record in the CCDB consists of a set of raw microscope images and any volumes, images, or data derived from it, along with a rich set of methodological details. These derived products include reconstructions, animations, correlated volumes, and the results of any segmentation or analysis performed on the data. By presenting all of the raw data, as well as reconstructed and processed data with a thorough description of how the specimen was prepared and imaged, researchers are free to extract additional content from micrographs that may not have been analyzed by the original author or employ additional alignment, reconstruction, or segmentation algorithms to the data.
The utility of image databases depends on the ability to query them on the basis of descriptive attributes and on their contents. Of these two types of query, querying images on the basis of their contents is by far the most challenging. Although the development of computer algorithms to identify and extract image features in image data is advancing,48 it is unlikely that any algorithm will be able to match the skill of an experienced microscopist for many years.
The CCDB project addresses this problem in two ways. One currently supported way is to store the results of segmentations and analyses performed by individual researchers on the data sets stored in the CCDB. The CCDB allows each object segmented from a reconstruction to be stored as a separate object in the database along with any quantitative information derived from it. The list of segmented objects and their morphometric quantities provides a means to query a dataset based on features contained in the data such as object name (e.g., dendritic spine) or quantities such as surface area, volume, and length.
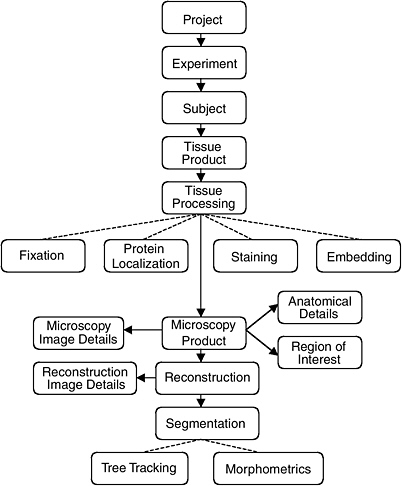
FIGURE 4.1 The schema and entity relationship in the Cell Centered Database.
SOURCE: See http://ncmir.ucsd.edu/CCDB.
It is also desirable to exploit information in the database that is not explicitly represented in the schema.49 Thus, the CCDB project team is developing specific data types around certain classes of segmented objects contained in the CCDB. For example, the creation of a “surface data type” will enable users to query the original surface data directly. The properties of the surfaces can be determined through very general operations at query time that allow the user to query on characteristics not explicitly modeled in the schema (e.g., dendrites from striatal medium spiny cells where the diameter of the dendritic shaft shows constrictions of at least 20 percent along its length). In this example, the schema does not contain explicit indication of the shape of the dendritic shaft, but these characteristics can be computed as part of the query processing. Additional data types are being developed for volume data and protein distribution data. A data type for tree structures generated by Neurolucida has recently been implemented.
The CCDB is being designed to participate in a larger, collaborative virtual data federation. Thus, an approach to reconciling semantic differences between various databases must be found.50 Scientific
terminology, particularly neuroanatomical nomenclature, is vast, nonstandard, and confusing. Anatomical entities may have multiple names (e.g., caudate nucleus, nucleus caudates), the same term may have multiple meanings (e.g., spine [spinal cord] versus spine [dendritic spine]), and worst of all, the same term may be defined differently by different scientists (e.g., basal ganglia). To minimize semantic confusion and to situate cellular and subcellular data from the CCDB in a larger context, the CCDB is mapped to several shared knowledge sources in the form of ontologies.
Concepts in the CCDB are being mapped to the Unified Medical Language System (UMLS), a large metathesaurus and knowledge source for the biomedical sciences.51 The UMLS assigns each concept in the ontology a unique identifier (ID); thus, all synonymous terms can then be assigned the same ID. For example, the UMLS ID number for the synonymous terms Purkinje cell, cerebellar Purkinje cell, and Purkinje’s corpuscle is C0034143. Thus, regardless of which term is preferred by a given individual, if they share the same ID, they are asserted to be the same. Conversely, even if two terms share the same name, they are distinguishable by their unique IDs. In the example given above, spine (spinal cord) = C0037949, whereas spine (dendritic spine) = C0872341.
In addition, an ontology can support the linkage of concepts by a set of relationships. These relationships may be simple “is a” and “has a” relationships (e.g., Purkinje cell is a neuron, neuron has a nucleus), or they may be more complex.52 From the above statements, a search algorithm could infer that “Purkinje cell has a nucleus” if the ontology is encoded in a form that would allow such reasoning to be performed. Because the knowledge required to link concepts is contained outside of the source database, the CCDB is relieved of the burden of storing exhaustive taxonomies for individual datasets, which may become obsolete as new knowledge is discovered.
The UMLS has recently incorporated the NeuroNames ontology53 as a source vocabulary. NeuroNames is a comprehensive resource for gross brain anatomy in the primate. However, for the type of cellular and subcellular data contained in the CCDB, the UMLS does not contain sufficient detail. Ontologies for areas such as neurocytology and neurological disease are being built on top of the UMLS, utilizing existing concepts wherever possible and constructing new semantic networks and concepts as needed.54
In addition, imaging data in the CCDB is mapped to a higher level of brain organization by registering their location in the coordinate system of a standard brain atlas. Placing data into an atlas-based coordinate systems provides one method by which data taken across scales and distributed across multiple resources can reliably be compared.55
Through the use of computer-based atlases and associated tools for warping and registration, it is possible to express the location of anatomical features or signals in terms of a standardized coordinate system. While there may be disagreement among neuroscientists about the identity of a brain area giving rise to a signal, its location in terms of spatial coordinates is at least quantifiable. The expression of brain data in terms of atlas coordinates also allows them to be transformed spatially to offer alternative views that may provide additional information (such as flat maps or additional parcellation
schemes).56 Finally, because individual experiments can study only a few aspects of a brain region at one time, a standard coordinate system allows the same brain region to be sampled repeatedly to allow data to be accumulated over time.
4.2.11 A Case Study: Ecological and Evolutionary Databases
Although genomic databases such as GenBank receive the majority of attention, databases and algorithms that operate on databases are key tools in research into ecology and biodiversity as well. These tools can provide researchers with access to information regarding all identified species of a given type, such as AlgaeBase57 or FishBase;58 they also serve as a repository for submission of new information and research. Other databases go beyond species listings to record individuals: for example, the ORNIS database of birds seeks to provide access to nearly 5 million individual specimens held in natural history collections, which includes data such as recordings of vocalizations and egg and nest holdings.59
The data associated with ecological research are gathered from a wide variety of sources: physical observations in the wild by both amateurs and professionals; fossils; natural history collections; zoos, botanical gardens, and other living collections; laboratories; and so forth. In addition, these data must placed into contexts of time, geographic location, environment, current and historical weather and climate, and local, regional, and global human activity. Needless to say, these data sources are scattered throughout many hundreds or thousands of different locations and formats, even when they are in digitally accessible format. However, the need for integrated ecological databases is great: only by being able to integrate the totality of observations of population and environment can certain key questions be answered. Such a facility is central to endangered species preservation, invasive species monitoring, wildlife disease monitoring and intervention, agricultural planning, and fisheries management, in addition to fundamental questions of ecological science.
The first challenge in building such a facility is to make the individual datasets accessible by networked query. Over the years, hundreds of millions of specimens have been recorded in museum records. In many cases, however, the data are not even entered into a computer; they may be stored as a set of index cards dating from the 1800s. Natural history collections, such as a museum’s collection of fossils, may not even be indexed, and they are available to researchers only by physically inspecting the drawers. Very few specimens have been geocoded.
Museum records carry a wealth of image and text data, and digitizing these records in a meaningful and useful way remains a serious challenge. For this reason, funding agencies such as the National Science Foundation (NSF) are emphasizing integrating database creation, curation, and sharing into the process of ecological science: for example, the NSF Biological Databases and Informatics program60 (which includes research into database algorithms and structures, as well as developing particular databases) and the Biological Research Collections program, which provides around $6 million per year for computerizing existing biological data. Similarly, the NSF Partnerships for Enhancing Expertise in Taxonomy (PEET) program,61 which emphasizes training in taxonomy, requires that recipients of funding incorporate collected data into databases or other shared electronic formats.
|
56 |
D.C. Van Essen, “Windows on the Brain: The Emerging Role of Atlases and Databases in Neuroscience,” Current Opinion in Neurobiology 12:574-579, 2002. |
|
57 |
|
|
58 |
|
|
59 |
|
|
60 |
NSF Program Announcement NSF 02-058; see http://www.nsf.gov/pubsys/ods/getpub.cfm?nsf02058. |
|
61 |
Ecological databases also rely on metadata to improve interoperability and compatibility among disparate data collections.62 Ecology is a field that demands access to large numbers of independent datasets such as geographic information, weather and climate records, biological specimen collections, population studies, and genetic data. These datasets are collected over long periods of time, possibly decades or even centuries, by a diverse set of actors for different purposes. A commonly agreed-upon format and vocabulary for metadata is essential for efficient cooperative access.
Furthermore, as data increasingly are collected by automated systems such as embedded systems and distributed sensor networks, the applications that attempt to fuse the results into formats amenable to algorithmic or human analysis must deal with high (and always on) data rates, likely contained in shifting standards for representation. Again, early agreement on a basic system for sharing metadata will be necessary for the feasibility of such applications.
In attempting to integrate or cross-query these data collections, a central issue is the naming of species or higher-level taxa. The Linnean taxonomy is the oldest such effort in biology, of course, yet because there is not yet (nor likely can ever be) complete agreement on taxa identification, entries in different databases may contain different tags for members of the same species, or the same tag for members that were later determined to be of different species. Taxa are often moved into different groups, split, or merged with others; names are sometimes changed. A central effort to manage this is the Integrated Taxonomic Information System (ITIS),63 which began life as a U.S. interagency task force, but today is a global cooperative effort between government agencies and researchers to arrive at a repository for agreed-upon species names and taxonomic categorization. ITIS data are of varying quality, and entries are tagged with three different quality indicators: credibility, which indicates whether or not data have been reviewed; latest review, giving the year of the last review; and global completeness, which records whether all species belonging to a taxon were included at the last review. These measurements allow researchers to evaluate whether the data are appropriate for their use.
In constructing such a database, many data standards questions arise. For example, ITIS uses naming standards from the International Code of Botanical Nomenclature and the International Code of Zoological Nomenclature. However, for the kingdom Protista, which at various times in biological science has been considered more like an animal and more like a plant, both standards might apply. Dates and date ranges provide another challenge: while there are many international standards for representing a calendar date, in general these did not foresee the need to represent dates occurring millions or billions of years ago. ITIS employs a representation for geologic ages, and this illustrates the type of challenge encountered when stretching a set of data standards to encompass many data types and different methods of collection.
For issues of representing observations or collections, an important element is the Darwin Core, a set of XML metadata standards for describing a biological specimen, including observations in the wild and preserved items in natural history collections. Where ITIS attempts to improve communicability by achieving agreement on precise name usage, Darwin Core64 (and similar metadata efforts) concentrates the effort on labeling and markup of data. This allows individual databases to use their own data structures, formats, and representations, as long as the data elements are labeled by Darwin Core keywords. Since the design demands on such databases will be substantially different, this is a useful approach. Another attempt to standardize metadata for ecological data is the Access to Biological Collections Data (ABCD) Schema,65 which is richer and contains more information. These two approaches indicate a common strategic choice: simpler standards are easier to adopt, and thus will likely be more widespread, but are limited in their expressiveness; more complex standards can successfully
|
62 |
For a more extended discussion of the issues involved in maintaining ecological data, see W.K. Michener and J.W. Brunt, eds., Ecological Data: Design, Management and Processing, Methods in Ecology, Blackwell Science, Maryland, 2000. A useful online presentation can be found at http://www.soest.hawaii.edu/PFRP/dec03mtg/michener.pdf. |
|
63 |
|
|
64 |
|
|
65 |
support a wider variety of queries and data types, but may be slower to gain adoption. Another effort to accomplish agreement on data and metadata standards is the National Biological Information Initiative (NBII), a program of the U.S. Geological Survey’s Center for Biological Informatics.
Agreement on standard terminology and data labeling would accomplish little if the data sources were unknown. The most significant challenge in creating large-scale ecological information is the integration and federation of the potentially vast number of relevant databases. The Global Biodiversity Information Facility (GBIF)66 is an attempt to offer a single-query interface to cooperating data providers; in December of 2004, it consisted of 95 providers totaling many tens of millions of individual records. GBIF accomplishes this query access through the use of data standards (such as the Darwin Core) and Web services, an information technology (IT) industry standard way of requesting information from servers in a platform-independent fashion. A similar international effort is found at the Clearinghouse Mechanism (CHM),67 an instrumentality of the Convention on Biodiversity. The CHM is intended as a way for information on biodiversity to be shared among signatory states and made available as a way to monitor compliance and as a tool for policy.
Globally integrated ecological databases are still in embryonic form, but as more data become digitized and made available by the Internet in standard fashions, their value will increase. Integration with phylogenetic and molecular databases will add to their value as research tools, in both the ecological and the evolutionary fields.
4.3 DATA PRESENTATION
4.3.1 Graphical Interfaces
Biological processes can take place over a vast array of spatial scales, from the nanoscale inhabited by individual molecules, to the everyday, meter-sized human world. They can take place over an even vaster range of time scales, from the nanosecond gyrations of a folding protein molecule to the seven decade (or so) span of a human life—and far beyond, if evolutionary time is included. They also can be considered at many levels of organization, from the straightforward realm of chemical interaction to the abstract realm of, say, signal transduction and information processing.
Much of 21st century biology must deal with these processes at every level and at every scale, resulting in data of high dimensionality. Thus, the need arises for systems that can offer vivid and easily understood visual metaphors to display the information at each level, showing the appropriate amount of detail. (Such a display would be analogous to, say, a circuit diagram, with its widely recognized icons for diodes, transistors, and other such components.) A key element of such systems is easily understood metaphors that present signals containing multiple colors over time on more than one axis. As an empirical matter, these metaphors are hard to find. Indeed, the problem of finding a visually (or intellectually!) optimal display layout for high-dimensional data is arguably combinatorially hard, because in the absence of a well-developed theory of display, it requires exploring every possible combination of data in a multitude of arrangements.
The system would likewise offer easy and intuitive ways to navigate between levels, so that the user could drill down to get more detail or pop up to higher abstractions as needed. Also, it would offer good ways to visualize the dynamical behavior of the system over time—whatever the appropriate time scale might be. Current-generation visualization systems such as those associated with BioSPICE68 and Cytoscape69 are a good beginning—but, as their developers themselves are the first to admit, only a beginning.
|
66 |
See http://www.gbif.org/. |
|
67 |
|
|
68 |
|
|
69 |
Biologists use a variety of different data representations to help describe, examine, and understand data. Biologists often use cartoons as conceptual, descriptive models of biological events or processes. A cartoon might show a time line of events: for example, the time line of the phosphorylation of a receptor that allows a protein to bind to it. As biologists take into account the simultaneous interactions of larger numbers of molecules, events over time become more difficult to represent in cartoons. New ways to “see” interactions and associations are therefore needed in life sciences research.
The most complex data visualizations are likely to be representations of networks. The complete graph in Figure 4.2 contains 4,543 nodes of approximately 6,000 proteins encoded by the yeast genome, along with 12,843 interactions. The graph was developed using the Osprey network visualization system.
FIGURE 4.2 From genomics to proteomics. Visualization of combined, large-scale interaction data sets in yeast. A total of 14,000 physical interactions obtained from the GRID database were represented with the Osprey network visualization system (see http://biodata.mshri.on.ca/grid). Each edge in the graph represents an interaction between nodes, which are colored according to Gene Ontology (GO) functional annotation. Highly connected complexes within the dataset, shown at the perimeter of the central mass, are built from nodes that share at least three interactions within other complex members. The complete graph contains 4,543 nodes of ~6,000 proteins encoded by the yeast genome, 12,843 interactions and an average connectivity of 2.82 per node. The 20 highly connected complexes contain 340 genes, 1,835 connections, and an average connectivity of 5.39.
SOURCE: Reprinted by permission from M. Tyers and M. Mann, “From Genomics to Proteomics,” Nature 422:193-197, 2003. Copyright 2003 Macmillan Magazines Ltd.
Other diagrammatic simulations of complex cell networks use tools such as the Diagrammatic Cell Language (DCL) and Visual Cell. These software tools are designed to read, query, and edit cell pathways, and to visualize data in a pathway context. Visual Cell creates detailed drawings by compactly formatting thousands of molecular interactions. The software uses DCL, which can visualize and simulate large-scale networks such as interconnected signal transduction pathways and the gene expression networks that control cell proliferation and apoptosis. DCL can visualize millions of chemical states and chemical reactions.
A second approach to diagrammatic simulation has been developed by Efroni et al.70 These researchers use the visual language of Statecharts, which makes specification of the simulation precise, legible, and machine-executable. Behavior in Statecharts is described by using states and events that cause transitions between states. States may contain substates, thus enabling description at multiple levels and zooming in and zooming out between levels. States may also be divided into orthogonal states, thus modeling concurrency, allowing the system to reside simultaneously in several different states. A cell, for example, may be described orthogonally as expressing several receptors, no receptors, or any combination of receptors at different stages of the cell cycle and in different anatomical compartments. Furthermore, transitions take the system from one state to another. In cell modeling, transitions are the result of biological processes or the result of user intervention. A biological process may be the result of an interaction between two cells or between a cell and various molecules. Statecharts provide a controllable way to handle the enormous dataset of cell behavior by enabling the separation of that dataset into orthogonal states and allowing transitions.
Still another kind of graphical interface is used for molecular visualization. Interesting biomolecules usually consist of thousands of atoms. A list of atomic coordinates is useful for some purposes, but an actual image of the molecule can often provide much more insight into its properties—and an image that can be manipulated (e.g., viewed from different angles) is even more useful. Virtual reality techniques can be used to provide the viewer with a large field of view, and to enable the viewer to interact with the virtual molecule and compare it to other molecules. However, many problems in biomolecular visualization tax the capability of current systems because of the diversity of operations required and because many operations do not fit neatly into the current architectural paradigm.
4.3.2 Tangible Physical Interfaces
As useful as graphical visualizations are, even in simulated three-dimensional virtual reality they are still two-dimensional. Tangible, physical models that a human being can manipulate directly with his or her hands are an extension of the two-dimensional graphical environment. A project at the Molecular Graphics Laboratory at the Scripps Research Institute is developing tangible interfaces for molecular biology.71 These interfaces use computer-driven autofabrication technology (i.e., three-dimensional printers) and result in physical molecular representations that one can hold in one’s hand.
These efforts have required the development and testing of software for the representation of physical molecular models to be built by autofabrication technologies, linkages between molecular descriptions and computer-aided design and manufacture approaches for enhancing the models with additional physical characteristics, and integration of the physical molecular models into augmented-reality interfaces as inputs to control computer display and interaction.
|
Box 4.4 Other methods [for the construction of large-scale topological maps of cellular networks] have sought to mine MEDLINE/PubMed abstracts that are considered to contain concise records of peer-reviewed published results. The simplest methods, often called ‘guilt by association,’ seek to find co-occurrence of genes or protein names in abstracts or even smaller structures such as sentences or phrases. This approach assumes that co-occurrences are indicative of functional links, although an obvious limitation is that negative relations (e.g., A does not regulate B) are counted as positive associations. To overcome this problem, other natural language processing methods involve syntactic parsing of the language in the abstracts to determine the nature of the interactions. There are obvious computation costs in these approaches, and the considerable complexity in human language will probably render any machine-based method imperfect. Even with limitations, such methods will probably be required to make knowledge in the extant literature accessible to machine-based analyses. For example, PreBIND used support vector machines to help select abstracts likely to contain useful biomolecular interactions to ‘backfill’ the BIND database. SOURCE: Reprinted by permission from J.J. Rice and G. Stolovitzky, “Making the Most of It: Pathway Reconstruction and Integrative Simulation Using the Data at Hand,” Biosilico 2(2):70-77. Copyright 2004 Elsevier. (References omitted.) |
4.3.3 Automated Literature Searching72
Still another form of data presentation is journal publication. It has not been lost on the scientific bioinformatics community that vast amounts of functional information that could be used to annotate gene and protein sequences are embedded in the written literature. Rice and Stolovitzky go so far as to say that mining the literature on biomolecular interactions can assist in populating a network model of intracellular interaction (Box 4.4).73
So far, however, the availability of full-text articles in digital formats such as PDF, HTML, or TIF files has limited the possibilities for computer searching and retrieval of full text in databases. In the future, wider use of structured documents tagged with XML will make intelligent searching of full text feasible, fast, and informative and will allow readers to locate, retrieve, and manipulate specific parts of a publication.
In the meantime, however, natural language provides a considerable, though not insurmountable, challenge for algorithms to extract meaningful information from natural text. One common application of natural language processing involves the extraction from the published literature of information about proteins, drugs, and other molecules. For example, Fukuda et al. (1998) pioneered identification of protein names using properties of the text such as the occurrence of uppercase letters, numerals, and special endings to pinpoint protein names.74
Other work has investigated the feasibility of recognizing interactions between proteins and other molecules. One approach is based on simultaneous occurrences of gene names and their use to predict their connections based on their occurrence statistics.75 A second approach to pathway discovery was
|
72 |
The discussion in Section 4.3.3 is based on excerpts from L. Hirschman, J.C. Park, J. Tsujii, L. Wong, and C.H. Wu, “Accomplishments and Challenges in Literature Data Mining for Biology,” Bioinformatics Review 18(12):1553-1561, 2002. Available at http://pir.georgetown.edu/pirwww/aboutpir/doc/data_mining.pdf. |
|
73 |
J.J. Rice and G. Stolovitzky, “Making the Most of It: Pathway Reconstruction and Integrative Simulation Using the Data at Hand,” Biosilico 2(2):70-77, 2004. |
|
74 |
K. Fukuda, et al., “Toward Information Extraction: Identifying Protein Names from Biological Papers,” Pacific Symposium on Biocomputing 1998, 707-718. (Cited in Hirschman et al., 2002.) |
|
75 |
B. Stapley and G. Benoit, “Biobibliometrics: Information Retrieval and Visualization from Co-occurrences of Gene Names in MEDLINE Abstracts,” Pacific Symposium on Biocomputing 2000, 529-540; J. Ding et al., “Mining MEDLINE: Abstracts, Sentences, or Phrases?” Pacific Symposium on Biocomputing 2002, 326-337. (Cited in Hirschman et al., 2002.) |
based on templates that matched specific linguistic structures to recognize and extract of protein interaction information from MEDLINE documents.76 More recent work goes beyond the analysis of single sentences to look at relations that span multiple sentences through the use of co-reference. For example, Putejovsky and Castano focused on relations of the word inhibit and showed that it was possible to extract biologically important information from free text reliably, using a corpus-based approach to develop rules specific to a class of predicates.77 Hahn et al. described the MEDSYNDIKATE system for acquiring knowledge from medical reports, a system capable of analyzing co-referring sentences and extracting new concepts given a set of grammatical constructs.78
Box 4.5 describes a number of other information extraction successes in biology. In a commentary in EMBO Reports on publication mining, Les Grivell, manager of the European electronic publishing initiative, E-BioSci, sums up the challenges this way:79
The detection of gene symbols and names, for instance, remains difficult, as researchers have seldom followed logical rules. In some organisms—the fruit fly Drosophila is an example—scientists have enjoyed applying gene names with primary meaning outside the biological domain. Names such as vamp, eve, disco, boss, gypsy, zip or ogre are therefore not easily recognized as referring to genes.80
Also, both synonymy (many different ways to refer to the same object) and polysemy (multiple meanings for a given word) cause problems for search algorithms. Synonymy reduces the number of recalls of a given object, whereas polysemy causes reduced precision. Another problem is ambiguities of a word’s sense. The word insulin, for instance, can refer to a gene, a protein, a hormone or a therapeutic agent, depending on the context. In addition, pronouns and definite articles and the use of long, complex or negative sentences or those in which information is implicit or omitted pose considerable hurdles for full-text processing algorithms.
Grivell points out that algorithms exist (e.g., the Vector Space Model) to undertake text analysis, theme generation, and summarization of computer-readable texts, but adds that “apart from the considerable computational resources required to index terms and to precompute statistical relationships for several million articles,” an obstacle to full-text analysis is the fact that scientific journals are owned by a large number of different publishers, so computational analysis will have to be distributed across multiple locations.
|
Box 4.5 Besides the recognition of protein interactions from scientific text, natural language processing has been applied to a broad range of information extraction problems in biology. Capturing of Specific Relations in Databases. … We begin with systems that capture specific relations in databases. Hahn et al. (2002) used natural language techniques and nomenclatures of the Unified Medical Language System (UMLS) to learn ontological relations for a medical domain. Baclawski et al. (2000) is a diagrammatic knowledge representation method called keynets. The UMLS ontology was used to build keynets. Using both domain-independent and domain-specific knowledge, keynets parsed texts and resolved references to build relationships between entities. Humphreys et al. (2000) described two information extraction applications in biology based on templates: EMPathIE extracted from journal articles details of enzyme and metabolic pathways; PASTA extracted the roles of amino acids and active sites in protein molecules. This work illustrated the importance of template matching, and applied the technique to terminology recognition. Rindflesch et al. (2000) described EDGAR, a system that extracted relationships between cancer-related drugs and genes from biomedical literature. EDGAR drew on a stochastic part-of-speech tagger, a syntactic parser able to produce partial parses, a rule-based system, and semantic information from the UMLS. The metathesaurus and lexicon in the knowledge base were used to identify the structure of noun phrases in MEDLINE texts. Thomas et al. (2000) customized an information extraction system called Highlight for the task of gathering data on protein interactions from MEDLINE abstracts. They developed and applied templates to every part of the texts and calculated the confidence for each match. The resulting system could provide a cost-effective means for populating a database of protein interactions. Information Retrieval and Clustering. The next papers [in this volume] focus on improving retrieval and clustering in searching large collections. Chang et al. (2001) modified PSI-BLAST to use literature similarity in each iteration of its search. They showed that supplementing sequence similarity with information from biomedical literature search could increase the accuracy of homology search result. Illiopoulos et al. (2001) gave a method for clustering MEDLINE abstracts based on a statistical treatment of terms, together with stemming, a “go-list,” and unsupervised machine learning. Despite the minimal semantic analysis, clusters built here gave a shallow description of the documents and supported concept discovery. Wilbur (2002) formalized the idea of a “theme” in a set of documents as a subset of the documents and a subset of the indexing terms so that each element of the latter had a high probability of occurring in all elements of the former. An algorithm was given to produce themes and to cluster documents according to these themes. Classification. … text processing has been used for classification. Stapley et al. (2002) used a support vector machine to classify terms derived by standard term weighting techniques to predict the cellular location of proteins from description in abstracts. The accuracy of the classifier on a benchmark of proteins with known cellular locations was better than that of a support vector machine trained on amino acid composition and was comparable to a handcrafted rule-based classifier (Eisenhaber and Bork, 1999). SOURCE: Reprinted by permission from L. Hirschman, J.C. Park, J. Tsujii, L. Wong, and C.H. Wu, “Accomplishments and Challenges in Literature Data Mining for Biology, Bioinformatics Review 18(12):1553-1561, 2002, available at http://pir.georgetown.edu/pirwww/aboutpir/doc/data_mining.pdf. Copyright 2002 Oxford University Press. |
4.4 ALGORITHMS FOR OPERATING ON BIOLOGICAL DATA
4.4.1 Preliminaries: DNA Sequence as a Digital String
The digital nature of DNA is a central evolutionary innovation for many reasons—that is, the “values” of the molecules making up the polymer are discrete and indivisible units. Just as an electronic digital computer abstracts various continuous voltage levels as 0 and 1, DNA abstracts a three-dimensional organization of atoms as A, T, G, and C. This has important biological benefits, including very high-accuracy replication, common and simplified ways for associated molecules to bind to sites, and low ambiguity in coding for proteins.
For human purposes in bioinformatics, however, the use of the abstraction of DNA as a digital string has had other equally significant and related benefits. It is easy to imagine the opposite case, in which DNA is represented as the three-dimensional locations of each atom in the macromolecule, and comparison of DNA sequences is a painstaking process of comparing the full structures. Indeed, this is very much the state of the art in representing proteins (which, although they can be represented as a digital string of peptides, are more flexible than DNA, so the digital abstraction leaves out the critically important features of folding). The digital abstraction includes much of the essential information of the system, without including complicating higher- and lower-order biochemical properties.81 The comparison of the state of the art in computational analysis of DNA sequences and protein sequences speaks in part to the enormous advantage that the digital string abstraction offers when appropriate.
The most basic feature of the abstraction is that it treats the arrangement of physical matter as information. An important advantage of this is that information-theoretic techniques can be applied to specific DNA strings or to the overall alphabet of codon-peptide associations. For example, computer science-developed concepts such as Hamming distance, parity, and error-correcting codes can be used to evaluate the resilience of information in the presence of noise and close alternatives.82
A second and very practical advantage is that as strings of letters, DNA sequences can be stored efficiently and recognizably in the same format as normal text.83 An entire human genome, for example, can be stored in about 3 gigabytes, costing a few dollars in 2003. More broadly, this means that a vast array of tools, software, algorithms, and software packages that were designed to operate on text could be adapted with little or no effort to operate on DNA strings as well. More abstract examples include the long history of research into algorithms to efficiently search, compare, and transform strings. For example, in 1974, an algorithm for identifying the “edit distance” of two strings was discovered,84 measuring the minimum number of changes, transpositions, and insertions necessary to transform one string into another. Although this algorithm was developed long before the genome era, it is useful to DNA analysis nonetheless.85
Finally, the very foundation of computational theory is the Turing machine, an abstract model of symbolic manipulation. Some very innovative research has shown that the DNA manipulations of some single-celled organisms are Turing-complete,86 allowing the application of a large tradition of formal language analysis to problems of cellular machinery.
|
81 |
A. Regev and E. Shapiro, “Cellular Abstractions: Cells as Computation,” Nature 419(6905): 343, 2002. |
|
82 |
D.A. MacDonaill, “A Parity Code Interpretation of Nucleotide Alphabet Composition,” Chemical Communications 18:2062-2063, 2002. |
|
83 |
Ideally, of course, a nucleotide could be stored using only two bits (or three to include RNA nucleotides as well). ASCII typically uses eight bits to represent characters. |
|
84 |
R.A. Wagner and M.J. Fischer, “The String-to-String Correction Problem,” Journal of the Association for Computing Machinery 21(1):168-173, 1974. |
|
85 |
See for example, American Mathematical Society, “Mathematics and the Genome: Near and Far (Strings),” April 2002. Available at http://www.ams.org/new-in-math/cover/genome5.html; M.S. Waterman, Introduction to Computational Biology: Maps, Sequences and Genomes, Chapman and Hall, London, 1995; M.S. Waterman, “Sequence Alignments,” Mathematical Methods for DNA Sequences, CRC, Boca Raton, FL, 1989, pp. 53-92. |
|
86 |
L.F. Landweber and L. Kari, “The Evolution of Cellular Computing: Nature’s Solution to a Computational Problem,” Biosystems 52(1-3):3-13, 1999. |
These comments should not be taken to mean that the abstraction of DNA into a digital string is cost-free. Although digital coding of DNA is central to the mechanisms of heredity, the nucleotide sequence cannot deal with nondigital effects that also play important roles in protein synthesis and function. Proteins do not necessarily bind only to one specific sequence; the overall proportions of AT versus CG in a region affect its rate of transcription; and the state of methylation of a region of DNA is an important mechanism for the epigenetic control of gene expression (and can indeed be inherited just as the digital code can be inherited).87 There are also numerous posttranslational modifications of proteins by processes such as acetylation, glycosylation, and phosphorylation, which by definition are not inherent in the genetic sequence.88 The digital abstraction also cannot accommodate protein dynamics or kinetics. Because these nondigital properties can have important effects, ignoring them puts a limit on how far the digital abstraction can support research related to gene finding and transcription regulation.
Last, DNA is often compared to a computer program that drives the functional behavior of a cell. Although this analogy has some merit, it is not altogether accurate. Because DNA specifies which proteins the cell must assemble, it is at least one step removed from the actual behavior of a cell, since the proteins—not the DNA—that determine (or at least have a great influence on) cell behavior.
4.4.2 Proteins as Labeled Graphs
A significant problem in molecular biology is the challenge of identifying meaningful substructural similarities among proteins. Although proteins, like DNA, are composed of strings made from a sequence of a comparatively small selection of types of component molecules, unlike DNA, proteins can exist in a huge variety of three-dimensional shapes. Such shapes can include helixes, sheets, and other forms generally referred to as secondary or tertiary structure.
Since the structural details of a protein largely determine its functions and characteristics, determining a protein’s overall shape and identifying meaningful structural details is a critical element of protein studies. Similar structure may imply similar functionality or receptivity to certain enzymes or other molecules that operate on specific molecular geometry. However, even for proteins whose three-dimensional shape has been experimentally determined through X-ray crystallography or nuclear magnetic resonance, finding similarities can be difficult due to the extremely complex geometries and large amount of data.
A rich and mature area of algorithm research involves the study of graphs, abstract representations of networks of relationships. A graph consists of a set of nodes and a set of connections between nodes called “edges.” In different types of graphs, edges may be one-way (a “directed graph”) or two-way (“undirected”), or edges may also have “weights” representing the distance or cost of the connection. For example, a graph might represent cities as nodes and the highways that connect them as edges weighted by the distance between the pair of cities.
Graph theory has been applied profitably to the problem of identifying structural similarities among proteins.89 In this approach, a graph represents a protein, with each node representing a single amino acid residue and labeled with the type of residue, and edges representing either peptide bonds or close spatial proximity. Recent work in this area has combined graph theory, data mining, and information theoretic techniques to efficiently identify such similarities.90
A significant computational aspect of this example is that since the general problem of identifying subgraphs is NP-complete,91 the mere inspiration of using graph theory to represent proteins is insufficient; sophisticated algorithmic research is necessary to develop appropriate techniques, data representations, and heuristics that can sift through the enormous datasets in practical times. Similarly, the problem involves subtle biological detail (e.g., what distance represents a significant spatial proximity, which amino acids can be classified together), and could not be usefully attacked by computer scientists alone.
4.4.3 Algorithms and Voluminous Datasets
Algorithms play an increasingly important role in the process of extracting information from large biological datasets produced by high-throughput studies. Algorithms are needed to search, sort, align, compare, contrast, and manipulate data related to a wide variety of biological problems and in support of models of biological processes on a variety of spatial and temporal scales. For example, in the language of automated learning and discovery, research is needed to develop algorithms for active and cumulative learning; multitask learning; learning from labeled and unlabeled data; relational learning; learning from large datasets; learning from small datasets; learning with prior knowledge; learning from mixed-media data; and learning causal relationships.92
The computational algorithms used for biological applications are likely to be rooted in mathematical and statistical techniques used widely for other purposes (e.g., Bayesian networks, graph theory, principal component analysis, hidden Markov models), but their adaptation to biological questions must address the constraints that define biological events. Because critical features of many biological systems are not known, algorithms must operate on the basis of working models and must frequently contend with a lack of data and incomplete information about the system under study (though sometimes simulated data suffices to test an algorithm). Thus, the results they provide must be regarded as approximate and provisional, and the performance of algorithms must be tested and validated by empirical laboratory studies. Algorithm development, therefore, requires the joint efforts of biologists and computer scientists.
Sections 4.4.4 through 4.4.9 describe certain biological problems and the algorithmic approaches to solving them. Far from giving a comprehensive description, these sections are intended to illustrate the complex substrate on which algorithms must operate and, further, to describe areas of successful and prolific collaboration between computer scientists and biologists.
Some of the applications described below are focused on identifying or measuring specific attributes, such as the identity of a gene, the three-dimensional structure of a protein, or the degree of genetic variability in a population. At the heart of these lines of investigation is the quest to understand biological function, (e.g., how genes interact, the physical actions of proteins, the physiological results of genetic differences). Further opportunities to address biological questions are likely to be as diverse as biology itself, although work on some of those questions is only nascent at this time.
4.4.4 Gene Recognition
Although the complete genomic sequences of many organisms have been determined, not all of the genes within those genomes have been identified. Difficulties in identifying genes from sequences of uncharacterized DNA stem mostly from the complexity of gene organization and architecture. Just a small fraction of the genome of a typical eukaryote consists of exons, that is, blocks of DNA that, when arranged according to their sequence in the genome, constitute a gene; in the human genome, the fraction is estimated at less than 3 percent.
Regions of the genome that are not transcribed from DNA into RNA include biological signals (such as promoters) that flank the coding sequence and regulate the gene’s transcription. Other untranscribed regions of unknown purpose are found between genes or interspersed within coding sequences.
Genes themselves can occasionally be found nested within one another, and overlapping genes have been shown to exist on the same or opposite DNA strands.93 The presence of pseudogenes (nonfunctional sequences resembling real genes), which are distributed in numerous copies throughout a genome, further complicates the identification of true protein-coding genes.94 Finally, it is known that most genes are ultimately translated into more than one protein through a process that is not completely understood. In the process of transcription, the exons of a particular gene are assembled into a single mature mRNA. However, in a process known as alternate splicing, various splicings omit certain exons, resulting in a family of variants (“splice variants”) in which the exons remain in sequence, but some are missing. It is estimated that at least a third of human genes are alternatively spliced,95 with certain splicing arrangements occurring more frequently than others. Protein splicing and RNA editing also play an important role. To understand gene structures completely, all of these sequence features have to be anticipated by gene recognition tools.
Two basic approaches have been established for gene recognition: the sequence similarity search, or lookup method, and the integrated compositional and signal search, or template method (also known as ab initio gene finding).96 Sequence similarity search is a well-established computational method for gene recognition based on the conservation of gene sequences (called homology) in evolutionarily related organisms. A sequence similarity search program compares a query sequence (an uncharacterized sequence) of interest with already characterized sequences in a public sequence database (e.g., databases of the Institute of Genomic Research (TIGR)97) and then identifies regions of similarity between the sequences. A query sequence with significant similarity to the sequence of an annotated (characterized) gene in the database suggests that the two sequences are homologous and have common evolutionary origin. Information from the annotated DNA sequence or the protein coded by the sequence can potentially be used to infer gene structure or function of the query sequence, including promoter elements, potential splice sites, start and stop codons, and repeated segments. Alignment tools, such as BLAST,98 FASTA, and Smith-Waterman, have been used to search for the homologous genes in the database.
Although sequence similarity search has been proven useful in many cases, it has fundamental limitations. Manning et al. note in their work on the protein kinase complement of the human genome
that although “all 518 [kinase] genes are covered by some EST [Expressed Sequence Tag] sequence, and ~90% are present in gene predictions from the Celera and public genome databases, … those predictions are often fragmentary or inaccurate and are frequently misannotated.”99
There are several reasons for these limitations. First, only a fraction of newly discovered sequences have identifiable homologous genes in the current databases.100 The proportion of vertebrate genes with no detectable similarity in other phyla is estimated to be about 50 percent,101 and this is supported by a recent analysis of human chromosome 22, where only 50 percent of the proteins are found to be similar to previously known proteins.102 Also, the most prominent vertebrate organisms in GenBank have only a fraction of their genomes present in finished (versus draft, error-prone) sequences. Hence, it is obvious that sequence similarity search within vertebrates is currently limited. Second, sequence similarity searches are computationally expensive when query sequences have to be matched against a large number of sequences in the databases.
To resolve this problem, a dictionary-based method, such as Identifier of Coding Exons (ICE), is often employed. In this method, gene sequences in the reference database are fragmented into subsequences of length k, and these subsequences make up the dictionary against which a query sequence is matched. If the subsequences corresponding to a gene have at least m consecutive matches with a query sequence, the gene is selected for closer examination. Full-length alignment techniques are then applied to the selected gene sequences. The dictionary-based approach significantly reduces the processing time (down to seconds per gene).
In compositional and signal search, a model (typically a hidden Markov model) is constructed that integrates coding statistics (measures indicative of protein coding functions) with signal detection into one framework. An example of a simple hidden Markov model for a compositional and signal search for a gene in a sequence sampled from a bacterial genome is shown in Figure 4.3. The model is first “trained” on sequences from the reference database and generates the probable frequencies of different nucleotides at any given position on the query sequence to estimate the likelihood that a sequence is in a different “state” (such as a coding region). The query sequence is predicted to be a gene if the product of the combined probabilities across the sequence exceeds a threshold determined by probabilities generated from sequences in the reference database.
The discussion above has presumed that biological understanding does not play a role in gene recognition. This is often untrue—gene-recognition algorithms make errors of omission and commission when run against genomic sequences in the absence of experimental biological data. That is, they fail to recognize genes that are present, or misidentify starts or stops of genes, or mistakenly insert or delete segments of DNA into the putative genes. Improvements in algorithm design will help to reduce these difficulties, but all the evidence to date shows that knowledge of some of the underlying science helps even more to identify genes properly.103
FIGURE 4.3 Hidden Markov model of a compositional signal and search approach for finding a gene in a bacterial genome.
The model has four features: (1) state of the sequence, of which four states are possible (coding, intergenic, start, and stop); (2) outputs, defined as the possible nucleotide(s) that can exist at any given state (A, C, T, G at coding and intergenic states; ATG and TAA at start and stop states, respectively); (3) emission possibilities—the probability that a given nucleotide will be generated in any particular state; and (4) transition probability (TP)—the probability that the sequence is in transition between two states.
To execute the model, emission and transition probabilities are obtained by training on the characterized genes in the reference database. The set of all possible combinations of states for the query sequence is then generated, and an overall probability for each combination of states is calculated. If the combination having the highest overall probability exceeds a threshold determined using gene sequences in the reference database, the query sequence is concluded to be a gene.
4.4.5 Sequence Alignment and Evolutionary Relationships
A remarkable degree of similarity exists among the genomes of living organisms.104 Information about the similarities and dissimilarities of different types of organisms presents a picture of relatedness between species (i.e., between reproductive groups), but also must provide useful clues to the importance, structure, and function of genes and proteins carried or lost over time in different species. “Comparative genomics” has become a new discipline within biology to study these relationships.
|
104 |
For example, 9 percent of E. coli genes, 9 percent of rice genes, 30 percent of yeast genes, 43 percent of mosquito genes, 75 percent of zebrafish genes, and 94 percent of rat genes have homologs in humans. See http://iubio.bio.Indiana.edu:8089/all/hgsummary.html (Summary Table August 2005). |
Alignments of gene and protein sequences from many different organisms are used to find diagnostic patterns to characterize protein families; to detect or demonstrate homologies between new sequences and existing families of sequences; to help predict the secondary and tertiary structures of new sequences; and to serve as an essential prelude to molecular evolutionary analysis.
To visualize relationships between genomes, evolutionary biologists develop phylogenetic trees that portray groupings of organisms, characteristics, genes, or proteins based on their common ancestries and the set of common characters they have inherited. One type of molecular phylogenetic tree, for example, might represent the amino acid sequence of a protein found in several different species. The tree is created by aligning the amino acid sequences of the protein in question from different species, determining the extent of differences between them (e.g., insertions, deletions, or substitutions of amino acids), and calculating a measure of relatedness that is ultimately reflected in a drawing of a tree with nodes and branches of different lengths.
The examination of phylogenetic relationships of sequences from several different species generally uses a method known as progressive sequence alignment, in which closely related sequences are aligned first, and more distant ones are added gradually to the alignment. Attempts at tackling multiple alignments simultaneously have been limited to small numbers of short sequences because of the computational power needed to resolve them. Therefore, alignments are most often undertaken in a stepwise fashion. The algorithm of one commonly used program (ClustalW) consists of three main stages. First, all pairs of sequences are aligned separately in order to calculate a distance matrix giving the divergence of each pair of sequences; second, a guide tree is calculated from the distance matrix; and third, the sequences are progressively aligned according to the branching order in the guide tree.
Alignment algorithms that test genetic similarity face several challenges. The basic premise of a multiple sequence alignment is that, for each column in the alignment, every residue from every sequence is homologous (i.e., has evolved from the same position in a common ancestral sequence). In the process of comparing any two amino acid sequences, the algorithm must place gaps or spaces at points throughout the sequences to get the sequences to align. Because inserted gaps are carried forward into subsequent alignments with additional new sequences, the cumulative alignment of multiple sequences can become riddled with gaps that sometimes result in an overall inaccurate picture of relationships between the proteins. To address this problem, gap penalties based on a weight matrix of different factors are incorporated into the algorithm. For example, the penalty for introducing a gap in aligning two similar sequences is greater that that for aligning two dissimilar sequences. Gap penalties differ depending on the length of the sequence, the types of sequence, and different regions of sequence. Based on the weight matrix and rules for applying penalties, the algorithm compromises in the placement of gaps to obtain the lowest penalty score for each alignment.
The placement of a gap in a protein sequence may represent an evolutionary change—if a gap, reflecting the putative addition or subtraction of an amino acid to a protein’s structure, is introduced, the function of the protein may change, and the change may have evolutionary benefit. However, the change may also be insignificant from a functional point of view. Today, it is known that most insertions and deletions occur in loops on the surface of the protein or between domains of multidomain proteins, which means that knowledge of the three-dimensional structure or the domain structure of the protein can be used to help identify functionally important deletions and insertions.
As the structures of different protein domains and families are increasingly determined by other means, alignment algorithms that incorporate such information should become more accurate. More recently, stochastic and iterative optimization methods are being used to refine individual alignments. Also, some algorithms (e.g., Bioedit) allow users to manually edit the alignment when other information or “eyeballing” suggests logical placement of gaps.
Exploitation of complete genomic knowledge across closely related species can play an important role in identifying the functional elements encoded in a genome. Kellis et al. undertook a comparative analysis of the yeast Saccharomyces cerevisiae based on high-quality draft sequences of three related
species (S. paradoxus, S. mikatae, and S. bayanus).105 This analysis resulted in significant revisions of the yeast gene catalogue, affecting approximately 15 percent of all genes and reducing the total count by about 500 genes. Seventy-two genome-wide elements were identified, including most known regulatory motifs and numerous new motifs, and a putative function was inferred for most of these motifs. The power of the comparative genomic approach arises from the fact that sequences that are positively selected (i.e., confer some evolutionary benefit or have some useful function) tend to be conserved as a species evolves, while other sequences are not conserved. By comparing a given genome of interest to closely related genomes, conserved sequences become much more obvious to the observer than if the functional elements had to be identified only by examination of the genome of interest. Thus, it is possible, at least in principle, that functional elements can be identified on the basis of conservation alone, without relying on previously known groups of co-regulated genes or without using data from gene expression or transcription factor binding experiments.
Molecular phylogenetic trees that graphically represent the differences between species are usually drawn with branch lengths proportional to the amount of evolutionary divergence between the two nodes they connect. The longer the distance between branches, the more relatively divergent are the sequences they represent. Methods for calculating phylogenetic trees fall into two general categories: (1) distance-matrix methods, also known as clustering or algorithmic methods, and (2) discrete data methods. In distance-matrix methods, the percentage of sequence difference (or distance) is calculated for pairwise combinations of all points of divergence; then the distances are assembled into a tree. In contrast, discrete data methods examine each column of the final alignment separately and look for the tree that best accommodates all of the information, according to optimality criteria—for example, the tree that requires the fewest character state changes (maximum parsimony), the tree that best fits an evolutionary model (maximum likelihood), or the tree that is most probable, given the data (Bayesian inference). Finally, “bootstrapping” analysis tests whether the whole dataset supports the proposed tree structure by taking random subsamples of the dataset, building trees from each of these, and calculating the frequency with which the various parts of the proposed tree are reproduced in each of the random subsamples.
Among the difficulties facing computational approaches to molecular phylogeny is the fact that some sequences (or segments of sequences) mutate more rapidly than others.106 Multiple mutations at the same site obscure the true evolutionary difference between sequences. Another problem is the tendency of highly divergent sequences to group together when being compared regardless of their true relationships. This occurs because of a background noise problem—with only a limited number of possible sequence letters (20 in the case of amino acid sequences), even divergent sequences will not infrequently present a false phylogenetic signal due strictly to chance.
4.4.6 Mapping Genetic Variation Within a Species
The variation that occurs between different species represents the product of reproductive isolation and population fission over very long time scales during which many mutational changes in genes and proteins occur. In contrast, variation within a single species is the result of sexual reproduction, genetic
recombination, and smaller numbers of relatively recent mutations.107 Examining the variation of gene or protein sequences between different species helps to draw a picture of the pedigree of a particular gene or protein over evolutionary time, but scientists are also interested in understanding the practical significance of such variation within a single species.
Geneticists have been trying for decades to identify the genetic variation among individuals in the human species that result in physical differences between them. There is an increasing recognition of the importance of genetic variation for medicine and developmental biology and for understanding the early demographic history of humans.108 In particular, variation in the human genome sequence is believed to play a powerful role in the origins of and prognoses for common medical conditions.109
The total number of unique mutations that might exist collectively in the entire human population is not known definitively and has been estimated at upward of 10 million,110 which in a 3 billion base-pair genome corresponds to a variant every 300 bases or less. Included in these are single-nucleotide polymorphisms (SNPs), that is, single-nucleotide sites in the genome where two or more of the four bases (A, C, T, G) occur in at least 1 percent of the population. Many SNPs were discovered in the process of overlapping the ends of DNA sequences used to assemble the human genome, when these sequences came from different individuals or from different members of a chromosome pair from the same individual. The average number of differences observed between the DNA of any two unrelated individuals represented at 1 percent or more in the population is one difference in every 1,300 bases; this leads to the estimation that individuals differ from one another at 2.4 million places in their genomes.111
In rare cases, a single SNP has been directly associated with a medical condition, such as sickle cell anemia or cystic fibrosis. However, most common diseases such as diabetes, cancer, stroke, heart disease, depression, and arthritis (to name a few) appear to have complex origins and involve the participation of multiple genes along with environmental factors. For this reason there is interest in identifying those SNPs occurring across the human genome that might be correlated with common medical conditions. SNPs found within exons that contain genes are of greatest interest because they are believed to be potentially related to changes in proteins that affect a predisposition to disease, but because most of the genome does not code for proteins (and indeed a number of noncoding SNPs have been found112), the functional impact of many SNPs is unknown.
Armed with rapid DNA sequencing tools and the ability to detect single-base differences, an international consortium looked for SNPs in individuals over the last several years, ultimately identifying more than 3 million unique SNPs and their locations on the genome in a public database. SNP maps of the human genome with a density of about one SNP per thousand nucleotides have been developed. An effort under way in Iceland known as deCODE seeks to correlate SNPs with human diseases.113 However, determining which combinations of the 10 million SNPs are associated with particular disease states, predisposition to disease, and genes that contribute to disease remains a formidable challenge.
Some research on this problem has recently on focused on the discovery that specific combinations of SNPs on a chromosome (called “haplotypes”) occur in blocks that are inherited together; that is, they
|
107 |
D. Posada and K.A. Crandall, “Intraspecific Gene Genealogies: Trees Grafting into Networks,” Trends in Ecology and Evolution 16(1):37-45, 2001. |
|
108 |
L.L. Cavalli-Sforza and M.W. Feldman, “The Application of Molecular Genetic Approaches to the Study of Human Evolution,” Nature Genetics 33 (Suppl.):266-275, 2003. |
|
109 |
S.B. Gabriel, S.F. Schaffner, H. Nguyen, J.M. Moore, J. Roy, B. Blumenstiel, J. Higins, et al., “The Structure of Haplotype Blocks in the Human Genome,” Science 296(5576):2225-2229, 2002. |
|
110 |
L. Kruglyak and D.A. Nickerson, “Variation Is the Spice of Life,” Nature Genetics 27(3):234-236, 2001, available at http://nucleus.cshl.edu/agsa/Papers/snp/Kruglyak_2001.pdf. |
|
111 |
The International SNP Map Working Group, “A Map of Human Genome Sequence Variation Containing 1.42 Million Single Nucleotide Polymorphisms,” Nature 409:928-933, 2001. |
|
112 |
See, for example, D. Trikka, Z. Fang, A. Renwick, S.H. Jones, R. Chakraborty, M. Kimmel, and D.L. Nelson, “Complex SNP-based Haplotypes in Three Human Helicases: Implications for Cancer Association Studies,” Genome Research 12(4):627-639, 2002. |
|
113 |
See www.decode.com. |
are unlikely to be separated by recombination that takes place during reproduction. Further, only a relatively small number of haplotype patterns appear across portions of a chromosome in any given population.114 This discovery potentially simplifies the problem of associating SNPs with disease because a much smaller number of “tag” SNPs (500,000 versus the estimated 10 million SNPs) might be used as representative markers for blocks of variation in initial studies to find correlations between parts of the genome and common diseases. In October 2002, the National Institutes of Health (NIH) launched the effort to map haplotype patterns (the HapMap) across the human genome.
Developing a haplotype map requires determination of all of the possible tag SNP combinations that are common in a population, and therefore relies on data from high-throughput screening of SNPs from a large number of individuals. A difficulty is that a haplotype represents a specific group of SNPs on a single chromosome. However, with the exception of gametes (sperm and egg), human cells contain two copies of each chromosome (one inherited from each parent). High-throughput studies generally do not permit the separate, parallel examination of each SNP site on both members of an individual’s pair of chromosomes. SNP data obtained from individuals represent a combination of information (referred to as the genotype) from both of an individual’s chromosomes. For example, genotyping an individual for the presence of a particular SNP will result in two data values (e.g., A and T). Each value represents an SNP at the same site on both chromosomes, and recently it has become possible to determine the specific chromosomes to which A and T belong.115
There are two problems in creating a HapMap. The first is to extract haplotype information computationally from genotype information for any individual. The second is to estimate haplotype frequencies in a population. Although good approaches to the first problem are known,116 the second remains challenging. Algorithms such as the expectation-maximization approach, Gibbs sampling method, and partition-ligation methods have been developed to tackle this problem.
Some algorithmic programs rely on the concept of evolutionary coalescence or a perfect phylogeny—that is, a rooted tree whose branches describe the evolutionary history of a set of sequences (or haplotypes) in sample individuals. In this scenario, each sequence has a single ancestor in the previous generation, under the presumption that the haplotype blocks have not been subject to recombination, and takes as a given that only one mutation will have occurred at any one SNP site. Given a set of genotypes, the algorithm attempts to find a set of haplotypes that fit a perfect phylogeny (i.e., could have originated from a common ancestor). The performance of algorithms for haplotype prediction generally improves as the number of individuals sampled and the number of SNPs included in the analysis increases. This area of algorithm development will continue to be a robust area of research in the future as scientists and industry seek to associate genetic variation with common diseases.
Direct haplotyping is also possible, and can circumvent many of the difficulties and ambiguities encountered when a statistical approach is used.117 For example, Ding and Cantor have developed a technique that enables direct molecular haplotyping of several polymorphic markers separated by as many as 24 kb.118 The haplotype is directly determined by simultaneously genotyping several polymorphic markers in the same reaction with a multiplex PCR and base extension reaction. This approach does not rely on pedigree data and does not require previous amplification of the entire genomic region containing the selected markers.
Finally, in early 2005, National Geographic and IBM announced a collaboration known as the the Genographic Project to probe the migratory history of the human species.119 The project seeks to collect 100,000 blood samples from indigenous populations, with the intent of analyzing DNA in these samples. Ultimately, the project will create a global database of human genetic variation and associated anthropological data (language, social customs, etc.) that provides a snapshot of human genetic variation before the cultural context of indigenous populations is lost—a context that is needed to make sense of the variations in DNA data.
4.4.7 Analysis of Gene Expression Data
Although almost all cells in an organism contain the same genetic material (the genomic blueprint for the entire organism), only about one-third of a given cell’s genes are expressed or “switched on”—that is, are producing proteins—at a given time. Expressed genes account for differences in cell types; for example, DNA in skin cells produces a different set of proteins than DNA in nerve cells. Similarly, a developing embryo undergoes rapid changes in the expression of its genes as its body structure unfolds. Differential expression in the same types of cells can represent different cellular “phenotypes” (e.g., normal versus diseased), and modifying a cell’s environment can result in changed levels of expression of a cell’s genes. In fact, the ability to perturb a cell and observe the consequential changes in expression is a key to understanding linkages between genes and can be used to model cell signaling pathways.
A powerful technology for monitoring the activity of all the genes in a cell is the DNA microarray (described in Box 7.5 in Chapter 7). Many different biological questions can be asked with microarrays, and arrays are now constructed in many varieties. For example, instead of DNA across an entire genome, the array might be spotted with a specific set of genes from an organism or with fabricated sequences of DNA (oligonucleotides) that might represent, for example, a particular SNP or a mutated form of a gene. More recently, protein arrays have been developed as a new tool that extends the reach of gene expression analysis.
The ability to collect and analyze massive sets of data about the transcriptional states of cells is an emerging focus of molecular diagnostics as well as drug discovery. Profiling the activation or suppression of genes within cells and tissues provides telling snapshots of function. Such information is critical not only to understand disease progression, but also to determine potential routes for disease intervention. New technologies that are driving the field include the creation of “designer” transcription factors to modulate expression, use of laser microdissection methods for isolation of specific cell populations, and technologies for capturing mRNA. Among the questions asked of microarrays (and the computational algorithms to decipher the results) are the discrimination of genes with significant changes in expression relative to the presence of a disease, drug regimen, or chemical or hormonal exposure.
To illustrate the power of large-scale analysis of gene data, an article in Science by Gaudet and Mango is instructive.120 A comparison of microarray data taken from Caenorhabditis elegans embryos lacking a pharynx with microarray data from embryos having excess pharyngeal tissue identified 240 genes that were preferentially expressed in the pharynx, and further identified a single gene as directly regulating almost all of the pharynx-specific genes that were examined in detail. These results suggest the possibility that direct transcriptional regulation of entire gene networks may be a common feature of organ-specification genes.121
|
119 |
More information on the project can be found at http://www5.nationalgeographic.com/genographic/. |
|
120 |
J. Gaudet and S.E. Mango, “Regulation of Organogenesis by the Caenorhabditis elegans FoxA Protein PHA-4,” Science 295(5556):821-825, 2002. |
|
121 |
For example, it is known that a specific gene activates other genes that function at two distinct steps of the regulatory hierarchy leading to wing formation in Drosophila (K.A. Guss, C.E. Nelson, A. Hudson, M. E. Kraus and S. B. Carroll, “Control of a Genetic Regulatory Network by a Selector Gene,” Science 292(5519):1164-1167, 2001), and also that the presence of specific factor is both necessary and sufficient for specification of eye formation in Drosophila imaginal discs, where it directly activates the expression of both early- and late-acting genes (W.J. Gehring and K. Ikeo, “Pax 6: Mastering Eye Morphogenesis and Evolution,” Trends in Genetics 15(9):371-377, 1999). |
Many analytic techniques have been developed and applied to the problem of revealing biologically significant patterns in microarray data. Various statistical tests (e.g., t-test, F-test) have been developed to identify genes with significant changes in expression (out of thousands of genes); such genes have had widespread attention as potential diagnostic markers or drug targets for disease, stages of development, and other cellular phenotypes. Many classification tools (e.g., Fisher’s Discriminant Analysis, Bayesian classifier, artificial neural networks, tools from signal processing) have also been developed to build a phenotype classifier with the genes differentially expressed. These classification tools are generally used to discriminate known sample groups from each other using differentially expressed genes selected by statistical testing.
Other algorithms are necessary because data acquired through microarray technology often have problems that must be managed prior to use. For example, the quality of microarray data is highly dependent on the way in which a sample is prepared. Many factors can affect the extent to which a dot fluoresces, of which the transcription level of the particular gene involved is only one. Such extraneous factors include the sample’s spatial homogeneity, its cleanliness (i.e., lack of contamination), the sensitivity of optical detectors in the specific instrument, varying hybridization efficiency between clones, relative differences between dyes, and so forth. In addition, because different laboratories (and different technicians) often have different procedures for sample preparation, datasets taken from different laboratories may not be strictly comparable. Statistical methods of analysis of variance (ANOVA) have been applied to deal with these problems, using models to estimate the various contributions to relative signal from the many potential sources. Importantly, these models not only allow researchers to attach measures of statistical significance to data, but also suggest improved experimental designs.122
An important analytical task is to identify groups of genes with similar expression patterns. These groups of genes are more likely to be involved in the same cellular pathways, and many data-driven hypotheses about cellular regulatory mechanisms (e.g., disease mechanisms) have been drawn under this assumption. For this purpose, various clustering methods, such as hierarchical clustering methods, self-organizing maps (trained neural networks), and COSA (Clustering Objects on Subsets of Attributes), have been developed. The goal of cluster analysis is to partition a dataset of N objects into subgroups such that these objects are more similar to those in their subgroups than to those in other groups. Clustering tools are generally used to identify groups of genes that have similar expression pattern across samples; thus, it is reasonable to suppose that the genes in each group (or cluster) are involved in the same biological pathway. Most clustering methods are iterative and involve the calculation of a notional distance between any two data points; this distance is used as the measure of similarity. In many implementations of clustering, the distance is a function of all of the attributes of each sample.
Agglomerative hierarchical clustering begins with assigning N clusters for N samples, where all samples are defined as different individual clusters. Potential clusters are arranged in a hierarchy displayed as a binary tree or “dendrogram.” Euclidian distance or Pearson correlation is used with “average linking” to develop the dendrogram. For example, two clusters that are closest to each other in terms of Euclidean distance are combined to form a new cluster, which is represented as the average of two groups combined (average linkage). This process is continued until there is one cluster to which all samples belong. In the process of forming the single cluster, the overall structure of clusters is evaluated for whether the merging of two clusters into one new cluster decreases both the sum of the similarity within all of the clusters and the sum of differences between all of the clusters. The clustering procedure stops at the level at which these are equal.
Self-organizing maps (SOMs)123 are another form of cluster analysis. With SOMs, a number of desired clusters is decided in advance, and a geometry of nodes (such as an N × M grid) is created, where each node represents a single cluster. The nodes are randomly placed in the data space. Then, in
a random order, each data point is selected. At each iteration, the nodes move closer to the selected data point, with the distance moved influenced by the distance from the data point to the node and the iteration number. Thus, the closest node will move the most. Over time, the initial geometry of the nodes will deform and each node will represent the center of an identified cluster. Experimentation is often necessary to arrive at a useful number of nodes and geometry, but since SOMs are computationally tractable, it is feasible to run many sessions. The properties of SOMs—partially structured, scalable to large datasets, unsupervised, easily visualizable—make them well suited for analysis of microarray data, and they have been used successfully to detect patterns of gene expression.124
In contrast to the above two methods, COSA is based on the assumption that better clustering can be achieved if only relevant genes are used in individual clusters. This is consistent with the idea of identifying differentially expressed genes (relevant genes) and then using only those genes to build a classifier. The search algorithm in COSA identifies an optimal set of variables that should be used to group individual clusters and which clusters should be merged when their similarity is assessed using the optimal set of variables identified. This idea was implemented by adding weights reflecting contributions of all genes to producing a particular set of sample clusters, and the search algorithm is then formulated as an optimization problem. The clustering results by COSA indicate that a subset of genes makes a greater contribution to a particular sample cluster than to other clusters.125
Clustering methods are being used in many types of studies. For example, they are particularly useful in modeling cell networks and in clustering disparate kinds of data (e.g., RNA data and non-RNA data; sequence data and protein data). Clustering can be applied to evaluate how feasible a given network structure is. Also, clustering is often combined with perturbation analysis to explore a set of samples or genes for a particular purpose. In general, clustering can be useful in any study in which local analyses with groups of samples or genes identified by clustering improve the understanding of the overall system.
Biclustering is an alternate approach to revealing meaningful patterns in the data.126 It seeks to identify submatrices in which the set of values has a low mean-squared residue, meaning that the each value is reasonably coherent with other members in its row and column. (However, excluding meaningless solutions with zero area, this problem is unfortunately NP-complete.) Advantages of this approach include that it can reveal clusters based on a subset of attributes, it simultaneously clusters genes with similar expression patterns and conditions with similar expression patterns, and most importantly, clusters can overlap. Since genes are often involved in multiple biological pathways, this can be used to reveal linkages that otherwise would be obscured by traditional cluster analysis.
While many analyses of microarray data consider a single snapshot in time, of course expression levels vary over time, especially due to the cellular life cycle. A challenge in analyzing microarray time-series data is that cell cycles may be unsynchronized, making it difficult to correctly identify correlations between data samples that have similar expression behavior. Statistical techniques can identify periodicity in series and look for phase-shifted correlations between pairs of samples,127 as well as more traditional clustering analysis.
A separate set of analytic techniques is referred to as supervised methods, in contrast to clustering and similar methods that run with no incoming assumptions. Supervised methods, in contrast, use existing knowledge of the dataset to classify data into one of a set of classes. In general, these techniques
|
124 |
P. Tamayo, D. Slonim, J. Mesirov, Q. Zhu, S. Kitareewwan, E. Dmitrovsky, E.S. Lander, and T.R. Golub, “Interpreting Patterns of Gene Expression with Self-organizing maps: Methods and Application to Hematopoietic Differentiation,” Proceedings of the National Academy of Sciences 96(6):2907-2912, 1999. |
|
125 |
J.H. Friedman and J.J. Meulman, “Clustering Objects on Subsets of Attributes,” Journal of the Royal Statistical Society Series B 66(4):815-849(34), 2004. |
|
126 |
Y. Cheng and G.M. Church, “Biclustering of Expression Data,” Proceedings of the Eighth International Conference on Intelligent Systems for Molecular Biology 8:93-103, 2000. |
|
127 |
V. Filkov, S. Skiena, and J. Zhi, “Analysis Techniques for Microarray Time-Series Data,” Journal of Computational Biology 9(2):317-330. Available at http://www.cs.ucdavis.edu/~filkov/papers/spellmananalysis.pdf. |
rely on training sets provided by the researchers, where the class membership of data is provided. Then, when presented with experimental data, supervised methods apply the learning from the training set to perform similar classifications. One such technique is support vector machines (SVMs), which are useful for highly multidimensional data. SVMs map the data into a “feature space” and then create (through one of a large number of possible algorithms) a hyperplane that separates the classes. Another common method is Artificial Neural Nets (see XREF), which train on a dataset with defined class membership; if the neural network classifies a member of the training set incorrectly, the error back-propagates through the system and updates the weightings. Unsupervised and supervised methods can be combined for “semisupervised” learning methods, in which heterogeneous training data can be both classified and unclassified.128
However, there is no analytic method optimal to any dataset. Thus, it would be useful to develop a scheme that can guide users to choose an appropriate method (e.g., in hierarchical clustering, an appropriate set of similarity measure, linkage method, and the measure used to determine the number of clusters) to achieve a reasonable analysis of their own datasets.
Ultimately, it is desirable to go beyond correlations and associations in the analysis of gene expression data to seek causal relationships. It is an elementary truism of statistics that indications of correlation are not by themselves indicators of causality—an experimental manipulation of one of more variables is always necessary to conclude a causal relationship. Nevertheless, analysis of microarray data can be helpful in suggesting experiments that might be particularly fruitful in uncovering causal relationships. Bayesian analysis allows one to make inferences about the possible structure of a genetic regulatory pathway on the basis of microarray data, but even advocates of such analysis recognize the need for experimental test. One work goes so far as to suggest that it is possible that automated processing of microarray data can suggest interesting experiments that will shed light on causal relationships, even if the existing data themselves don’t support causal inferences.129
4.4.8 Data Mining and Discovery
4.4.8.1 The First Known Biological Discovery from Mining Databases130
By the early 1970s, the simian sarcoma virus had been determined to cause cancer in certain species of monkeys. In 1983, the responsible oncogene within the virus was sequenced. At around the same time, and entirely independently, a partial amino acid sequence of an important growth factor in humans—the platelet-derived growth factor (PDGF) was also determined. PDGF was known to cause cultured cells to proliferate in a cancer-like manner. Russell Doolittle compared the two sequences and found a high degree of similarity between them, indicating a possible connection between an oncogene and a normal human gene. In this case, the indication was that the simian sarcoma virus acted on cells in monkeys in a manner similar to the action of PDGF on human cells.
|
128 |
T. Li, S. Zhu, Q. Li, and M. Ogihara, “Gene Functional Classification by Semisupervised Learning from Heterogeneous Data,” pp. 78-82 in Proceedings of the ACM Symposium on Applied Computing, ACM Press, New York, 2003. |
|
129 |
C. Yoo and G. Cooper, “An Evaluation of a System That Recommends Microarray Experiments to Perform to Discover Gene-regulation Pathways,” Artificial Intelligence in Medicine 31(2):169-182, 2004, available at http://www.phil.cmu.edu/projects/genegroup/papers/yoo2003a.pdf. |
|
130 |
Adapted from S.G.E. Andersson and L. Klasson, “Navigating Through the Databases,” available at http://artedi.ebc.uu.se/course/overview/navigating_databases.html. The original Doolittle article was published as R.F. Doolittle, M.W. Hunkapiller, L.E. Hood, S.G. Davare, K.C. Robbins, S.A. Aaronson, and H.N. Antoniades, “Simian Sarcoma Virus onc Gene, v-sis, Is Derived from the Gene (or Genes) Encoding a Platelet-derived Growth Factor,” Science 221(4607):275-277, 1983. |
4.4.8.2 A Contemporary Example: Protein Family Classification and Data Integration for Functional Analysis of Proteins
New bioinformatics methods allow inference of protein function using associative analysis (“guilt by association”) of functional properties to complement the traditional sequence homology-based methods.131 Associative properties that have been used to infer function not evident from sequence homology include co-occurrence of proteins in operons or genome context; proteins sharing common domains in fusion proteins; proteins in the same pathway, subcellular network, or complex; proteins with correlated gene or protein expression patterns; and protein families with correlated taxonomic distribution (common phylogenetic or phyletic patterns).
Coupling protein classification and data integration allows associative studies of protein family, function, and structure.132 An example is provided in Figure 4.4, which illustrates how the collective use of protein family, pathway, and genome context in bacteria helped researchers to identify a long-sought human gene associated with the methylmalonic aciduria disorder.
Domain-based or structural classification-based searches allow identification of protein families sharing domains or structural fold classes. Functional convergence (unrelated proteins with the same activity) and functional divergence are revealed by the relationships between the enzyme classification and protein family classification. With the underlying taxonomic information, protein families that occur in given lineages can be identified. Combining phylogenetic pattern and biochemical pathway information for protein families allows identification of alternative pathways to the same end product in different taxonomic groups, which may present attractive potential drug targets. The systematic approach for protein family curation using integrative data leads to novel prediction and functional inference for uncharacterized “hypothetical” proteins, and to detection and correction of genome annotation errors (a few examples are listed in Table 4.2). Such studies may serve as a basis for further analysis of protein functional evolution, and its relationship to the coevolution of metabolic pathways, cellular networks, and organisms.
Underlying this approach is the availability of resources that provide analytical tools and data. For example, the Protein Information Resource (PIR) is a public bioinformatics resource that provides an advanced framework for comparative analysis and functional annotation of proteins. PIR recently joined the European Bioinformatics Institute and Swiss Institute of Bioinformatics to establish UniProt,133 an international resource of protein knowledge that unifies the PIR, Swiss-Prot, and TrEMBL databases. Central to the PIR-UniProt functional annotation of proteins is the PIRSF (SuperFamily) classification system134 that provides classification of whole proteins into a network structure to reflect their evolutionary relationships. This framework is supported by the iProClass integrated database of protein family, function, and structure,135 which provides value-added descriptions of all UniProt proteins with rich links to more than 50 other databases of protein family, function, pathway, interaction, modification, structure, genome, ontology, literature, and taxonomy. As a core resource, the PIR environment is widely used by researchers to develop other bioinformatics infrastructures and algorithms and to enable basic and applied scientific research, as shown by examples in Table 4.3.
FIGURE 4.4 Integration of protein family, pathway, and genome context data for disease gene identification.
The ATR enzyme (EC 2.5.1.17) converts inactive cobalamins to AdoCbl (A), a cofactor for enzymes in several pathways, including diol/glycerol dehydratase (EC 4.2.1.28) (B) and methylmalonyl-CoA mutase (MCM) (EC 5.4.99.2) (C). Many prokaryotic ATRs are predicted to be required for EC 4.2.1.28 based on the genome context of the corresponding genes. However, in at least one organism (Archaeoglobus fulgidus), the ATR gene is adjacent to the MCM gene, which provided a clue for cloning the human and bovine ATRs.
SOURCE: Courtesy of Cathy Wu, Georgetown University.
TABLE 4.2 Protein Family Classification and Integrative Associative Analysis for Functional Annotation
4.4.9 Determination of Three-dimensional Protein Structure
One central problem of proteomics is that of protein folding. Protein folding is one of the most important cellular processes because it produces the final conformation required for a protein to attain biological activity. Diseases such as Alzheimer’s disease or bovine spongiform encephalopathy (BSE, or “Mad Cow” disease) are associated with the improper folding of proteins. For example, in BSE the protein (called the scrapie prion), which is soluble when it folds properly, becomes insoluble when one of the intermediates along its folding pathway misfolds and forms an aggregation that damages nerve cells.136
Due to the importance of the functional conformation of proteins, many efforts have been attempted to predict computationally a three-dimensional structure of a protein from its amino acid sequence. Although experimental determination of protein structure based on X-ray crystallography and nuclear magnetic resonance yields protein structures in high resolution, it is slow, labor-intensive, and expensive and thus not appropriate for large-scale determination. Also, it can apply only to already-synthesized or isolated proteins, while an algorithm could be used to predict the structure of a great number of potential proteins.
TABLE 4.3 Algorithms, Databases, Analytical Systems, and Scientific Research Enabled by the PIR Resource
|
Resource |
Topic |
Reference |
|
Algorithm |
Benchmarking for sequence similarity search statistics |
Pearson, J. Mol. Biol. 276:71-84, 1998 |
|
PANDORA keyword-based analysis of proteins |
Kaplan, Nucleic Acids Research 31:5617-5626, 2003 |
|
|
Computing motif correlations for structure prediction |
Horng et al., J. Comp. Chem. 24(16):2032-2043, 2003 |
|
|
Database |
NESbase database of nuclear export signals |
la Cour et al., Nucleic Acids Research 31(l):393-396, 2003 |
|
TMPDB database of transmembrane topologies |
Ikeda et al., Nucleic Acids Research 31:406-409, 2003 |
|
|
SDAP database and tools for allergenic proteins |
Ivanciuc et al., Nucleic Acids Research 31:359-362, 2003 |
|
|
System |
SPINE 2 system for collaborative structural proteomics |
Goh et al., Nucleic Acids Research 31:2833-2838, 2003 |
|
ERGOTM genome analysis and discovery system |
Overbeek et al., Nucleic Acids Research 31(l):164-171, 2003 |
|
|
Automated annotation pipeline and cDNA annotation system |
Kasukawa et al., Genome Res. 13(6B):1542-1551, 2003 |
|
|
Systers, GeneNest, SpliceNest from genome to protein |
Krause et al., Nucleic Acids Research 30(l):299-300, 2002 |
|
|
Research |
Intermediate filament proteins during carcinogenesis or apoptosis |
Prasad et al., Int. J. Oncol. 14(3):563-570, 1999 |
|
Conserved pathway by global protein network alignment |
Kelley et al., PNAS 100(20):11394-11399, 2003 |
|
|
Membrane targeting of phospholipase C pleckstrin |
Singh and Murray, Protein Sci. 12:1934-1953, 2003 |
|
|
Analysis of human and mouse cDNA sequences |
Strausberg et al., PNAS 99(26):16899-16903, 2002 |
|
|
A novel Schistosoma mansoni G protein-coupled receptor |
Hamdan et al., Mol. Biochem. Parasitol. 119(l):75-86, 2002 |
|
|
Proteomics reveals open reading frames (ORFs) in Mycobacterium tuberculosis |
Jungblut et al., Infect. Immunol. 69(9):5905-5907, 2001 |
Protein structures predicted in high resolution can help characterize the biological functions of proteins. Biotechnology companies are hoping to accelerate their efforts to discover new drugs that interact with proteins by using structure-based drug design technologies. By combining computational and combinatorial chemistry, researchers expect to find more viable leads. Algorithms create molecular structure built de novo to optimize interactions within the protein’s active sites. The use of so-called virtual screening in combination with studies of co-crystallized drugs and proteins could be a powerful tool for drug development.
A number of tools for protein structure prediction have been developed, and progress in prediction by these methods has been evaluated by the Critical Assessment of Protein Structure Prediction (CASP) experiment held every two years since 1994.137 In a CASP experiment, the amino acid sequences of proteins whose experimentally determined structures have not yet been released are published, and computational research groups are then invited to predict structures of these target sequences using their methods and any other publicly available information (e.g., known structures that exist in the Protein Data Bank (PDB), the data repository for protein structures). The methods used by the groups
can be divided into three areas depending on the similarity of the target protein to proteins of known structure: comparative (also known as homology) modeling, fold recognition (also known as threading), and de novo/new fold methods (also known as ab initio). This traditional division of prediction methods has become blurred as the methods in each category incorporate detailed information used by methods in the other categories.
In comparative (or homology) modeling, one or more template proteins of known structure with high sequence homology (greater than 25 percent sequence identity) to the target sequence are identified. The target and template sequences are aligned through multiple sequence alignment (similar to comparative genomics), and a three-dimensional structure of the target protein is generated from the coordinates of the aligned residues of the template proteins. Finally, the model is evaluated using a variety of criteria, and if necessary, the alignment and the three-dimensional model are refined until a satisfactory model is obtained.
If no reliable template protein can be identified from sequence homology alone, the prediction problem is denoted as a fold recognition (or threading) problem. The primary goal is to identify one or more folds in the template proteins that are consistent with the target sequence. In the classical threading methods, known as “rigid body assembly,” a model is constructed from a library of known core regions, loops, side chains, and folds, and the target sequence is then threaded onto the known folds. After evaluating how well the model fits the known folds, the best fit is chosen. The assumption in fold recognition is that only a finite number of folds exist and most existing folds can be identified from known structures in the PDB. Indeed, as new sequences are deposited and more protein structures are solved, there appear to be fewer and fewer unique folds. When two sequences share more than 25 percent similarity (or sequence identity), their structures are expected to have similar folds. However, there are still remaining issues such as the high rate of false positives in fold recognition, and therefore, the resulting alignment with the fold structure is poor. At 30 percent sequence identity, the fraction of incorrectly aligned residues is about 20 percent, and the number rises sharply with further decreases in sequence similarity. This limits the usefulness of comparative modeling.138
If no template structure (or fold) can be identified with confidence by sequence homology methods, the target sequence may be modeled using new fold prediction methods. The goal in this prediction method rests on the biological assumption that proteins adopt their lowest free energy conformation as their functional state. Thus, computational methods to predict structure ab initio comprise three elements: (1) protein geometry, (2) potential energy functions, and (3) an energy space search method (energy minimization method). First, setting protein geometry involves determining the number of particles to be used to represent the protein structure (for example, all-atom, united-atom, or virtual-atom model) and the nature of the space where atoms can be allocated (e.g., continuous (off-lattice) or discrete (lattice) model). In a simple ab initio folding such as a virtual-atom lattice model, one virtual atom represents a number of atoms in a protein (i.e., the backbone is represented as a sequence of alpha carbons) and an optimization method searches only the predetermined lattice points for positions of the virtual atoms to minimize the energy functions. Second, the potential energy functions in ab initio models include covalent terms, such as bond stretching, bond angle stretching, improper dihedrals, and torsional angles, and noncovalent terms, such as electrostatic and van der Waals forces. The use of molecular mechanics for refinement in comparative modeling is equivalent to ab initio calculation using all atoms in an off-lattice model. Third, many optimizations tools, such as genetic algorithms, Monte Carlo, simulated annealing, branch and bound, and successive quadratic programming (SQP), have been used to search for the global minimum in the energy (or structure) spaces with a number of local minima. These approaches have provided encouraging results, although the performance of each method may be limited by the shape of the energy space.
Beyond studies of protein structure is the problem of describing a solvent environment (such as water) and its influence on a protein’s conformational behavior. The importance of hydration in protein stability and folding is widely accepted. Models are needed to incorporate the effects of solvents in protein three-dimensional structure.
4.4.10 Protein Identification and Quantification from Mass Spectrometry
A second important problem in proteomics is protein identification and quantification. That is, given a particular biological sample, what specific proteins are present and in what quantities? This problem is at the heart of studying protein–protein interactions at proteomic scale, mapping various organelles, and generating quantitative protein profiles from diverse species. Making inferences about protein identification and abundance in biological samples is often challenging, because cellular proteomes are highly complex and because the proteome generally involves many proteins at relatively low abundances. Thus, highly sensitive analytical techniques are necessary.
Today, techniques based on mass spectrometry increasingly fill this need. The mass spectrometer works on a biological sample in ionized gaseous form. A mass analyzer measures the mass-to-charge ratio (m/z) of the ionized analytes, and a detector measures the number of ions at each m/z value. In the simplest case, a procedure known as peptide mass fingerprinting (PMF) is used. PMF is based on the fact that a protein is composed of multiple peptide groups, and identification of the complete set of peptides will with high probability characterize the protein in question. After enzymatically breaking up the protein into its constituent peptides, the mass spectrometer is used to identify individual peptides, each of which has a known mass. The premise of PMF is that only a very few (one in the ideal case) proteins will correspond to any particular set of peptides, and protein identification is effected by finding the best fit of the observed peptide masses to the calculated masses derived from, say, a sequence database. Of course, the “best fit” is an algorithmic issue, and a variety of approaches have been taken to determine the most appropriate algorithms.
The applicability of PMF is limited when samples are complex (that is, when they involve large numbers of proteins at low abundances). The reason is that only a small fraction of the constituent peptides are typically ionized, and those that are observed are usually from the dominant proteins in the mixture. Thus, for complex samples, multiple (tandem) stages of mass spectrometry may be necessary. In a typical procedure, peptides from a database are scored on the likelihood of their generating a tandem mass spectrum, and the top scoring peptide is chosen. This computational approach has shown great success, and contributed to the industrialization of proteomics.
However, much remains to be done. First, the generation of the spectrum is a stochastic process governed by the peptide composition, and the mass spectrometer. By mining data to understand these fragmentation propensities, scoring and identification can be further improved. Second, if the peptide is not in the database, de novo or homology-based methods must be developed for identification. Many proteins are post-translationally modified, with the modifications changing the mass composition. Enumeration and scoring of all modifications leads to a combinatorial explosion that must be addressed using novel computational techniques. It is fair to say that computation will play an important role in the success of mass spectrometry as the tool of choice for proteomics.
Mass spectrometry is also coming into its own for protein expression studies. The major problem here is that the intensity of a peak depends not only on the peptide abundance, but also on the physicochemical properties of the peptide. This makes it difficult to measure expression levels directly. However, relative abundance can be measured using the proven technique of stable-isotope dilution. This method makes use of the facts that pairs of chemically identical analytes of different stable-isotope composition can be differentiated in a mass spectrometer owing to their mass difference, and that the ratio of signal intensities for such analyte pairs accurately indicates the abundance ratio for the two analytes.
This approach shows great promise. However, computational methods are needed to correlate data across different experiments. If the data were produced using liquid chromatography coupled with
mass spectrometry, a peptide pair could be approximately labeled by its retention time in the column, and its mass-to-charge ratio. Such pairs can be matched across experiments using geometric matching. Combining the relative abundance levels from different experiments using statistical methods will greatly help in improving the reliability of this approach.
4.4.11 Pharmacological Screening of Potential Drug Compounds139
The National Cancer Institute (NCI) has screened more than 60,000 compounds against a panel of 60 human cancer cell lines. The extent to which any single compound inhibits growth in any given cell line is simply one data point relevant to that compound-cell line combination—namely the concentration associated with a 50 percent inhibition in the growth of that cell line. However, the pattern of such values across all 60 cell lines can provide insight into the mechanisms of drug action and drug resistance. Combined with molecular structure data, these activity patterns can be used to explore the NCI database of 460,000 compounds for growth-inhibiting effects in these cell lines, and can also provide insight into potential target molecules and modulators of activity in the 60 cell lines. Based on this approach, five compounds have been screened in this manner and selected for entry into clinical trials.
This approach to drug discovery and molecular pharmacology serves a number of useful functions. According to Weinstein et al.,
-
It suggests novel targets and mechanisms of action or modulation.
-
It detects inhibition of integrated biochemical pathways not adequately represented by any single molecule or molecular interaction. (This feature of cell-based assays is likely to be more important in the development of therapies for cancer than it is for most other diseases; in the case of cancer, one is fighting the plasticity of a poorly controlled genome and the selective evolutionary pressures for development of drug resistance.)
-
It provides candidate molecules for secondary testing in biochemical assays; conversely, it provides a well-characterized biological assay in vitro for compounds emerging from biochemical screens.
-
It ‘‘fingerprints’’ tested compounds with respect to a large number of possible targets and modulators of activity.
-
It provides such fingerprints for all previously tested compounds whenever a new target is assessed in many or all of the 60 cell lines. (In contrast, if a battery of assays for different biochemical targets were applied to, for example, 60,000 compounds, it would be necessary to retest all of the compounds for any new target or assay.)
-
It links the molecular pharmacology with emerging databases on molecular markers in microdissected human tumors—which, under the rubric of this article, constitute clinical (C) databases.
-
It provides the basis for pharmacophore development and searches of an S [structure] database for additional candidates. If an agent with a desired action is already known, its fingerprint patterns of activity can be used by … [various] pattern-recognition technologies to find similar compounds.
Box 4.6 provides an example of this approach.
4.4.12 Algorithms Related to Imaging
Biological science is rich in images. Most familiar are images taken through optical microscopes, but there are many other imaging modalities—electron microscopes, computed tomography scans, X-rays, magnetic resonance imaging, and so on. For most of the history of life science research, images have
|
139 |
Section 4.4.11 is based heavily on J.N. Weinstein, T.G. Myers, P.M. O’Connor, S.H. Friend, A.J. Fornace, Jr., K.W. Kohn, T. Fojo, et al., “An Information-Intensive Approach to the Molecular Pharmacology of Cancer,” Science 275(5298):343-349, 1997. |
|
Box 4.6 Given one compound as a “seed,” [an algorithm known as] COMPARE searches the database of screened agents for those most similar to the seed in their patterns of activity against the panel of 60 cell lines. Similarity in pattern often indicates similarity in mechanism of action, mode of resistance, and molecular structure…. A formulation of this approach in terms of three databases [includes databases for] the activity patterns [A], … molecular structural features of the tested compounds [S], and … possible targets or modulators of activity in the cells [T]…. The (S) database can be coded in terms of any set of two-dimensional (2D) or 3D molecular structure descriptors. The NCI’s Drug Information System (DIS) contains chemical connectivity tables for approximately 460,000 molecules, including the 60,000 tested to date. 3-D structures have been obtained for 97% of the DIS compounds, and a set of 588 bitwise descriptors has been calculated for each structure by use of the Chem-X computational chemistry package. This data set provides the basis for pharmacophoric searches; if a tested compound, or set of compounds, is found to have an interesting pattern of activity, its structure can be used to search for similar molecules in the DIS database. In the target (T) database, each row defines the pattern (across 60 cell lines) of a measured cell characteristic that may mediate, modulate, or otherwise correlate with the activity of a tested compound. When the term is used in this general shorthand sense, a “target” may be the site of action or part of a pathway involved in a cellular response. Among the potential targets assessed to date are oncogenes, tumor-suppressor genes, drug resistance-mediating transporters, heat shock proteins, telomerase, cytokine receptors, molecules of the cell cycle and apoptotic pathways, DNA repair enzymes, components of the cytoarchitecture, intracellular signaling molecules, and metabolic enzymes. In addition to the targets assessed one at a time, others have been measured en masse as part of a protein expression database generated for the 60 cell lines by 2D polyacrylamide gel electrophoresis. Each compound displays a unique “fingerprint” pattern, defined by a point in the 60D space (one dimension for each cell line) of possible patterns. In information theoretic terms, the transmission capacity of this communication channel is very large, even after one allows for experimental noise and for biological realities that constrain the compounds to particular regions of the 60D space. Although the activity data have been accumulated over a 6-year period, the experiments have been reproducible enough to generate … patterns of coherence. SOURCE: Reprinted by permission from J.N. Weinstein, T.G. Myers, P.M. O’Connor, S.H. Friend, A.J. Fornace, Jr., K.W. Kohn, T. Fojo, et al., “An Information-intensive Approach to the Molecular Pharmacology of Cancer,” Science 275(5298):343-349, 1997. Copyright 1997 AAAS. |
been a source of qualitative insight.140 While this is still true, there is growing interest in using image data more quantitatively.
Consider the following applications:
-
Automated identification of fungal spores in microscopic digital images and automated estimation of spore density;141
-
Automated analysis of liver MRI images from patients with putative hemochromatosis to determine the extent of iron overload, avoiding the need for an uncomfortable liver biopsy;142
|
140 |
Note also that biological imaging itself is a subset of the intersection between biology and visual techniques. In particular, other biological insight can be found in techniques that consider spectral information, e.g., intensity as a function of frequency and perhaps a function of time. Processing microarray data (discussed further in Section 7.2.1) ultimately depends on the ability to extract interesting signals from patterns of fluorescing dots, as does quantitative comparison of patterns obtained in two-dimensional polyacrylamide gel electrophoresis. (See S. Veeser, M.J. Dunn, and G.Z. Yang, “Multiresolution Image Registration for Two-dimensional Gel Electrophoresis,” Proteomics 1(7):856-870, 2001, available at http://vip.doc.ic.ac.uk/2d-gel/2D-gel-final-revision.pdf.) |
|
141 |
T. Bernier and J.A. Landry, “Algorithmic Recognition of Biological Objects,” Canadian Agricultural Engineering 42(2):101-109, 2000. |
|
142 |
George Reeke, Rockefeller University, personal communication to John Wooley, October 8, 2004. |
|
Box 4.7 Responding to the need to manage a large number of multispectral movies of mitotic cells in the late 1990s, Sorger and Swedlow began work on the open microscopy environment (OME). The OME is designed as infrastructure that manages optical microscopy images, storing both the primary image data and appropriate metadata on those images, including data on the optics of the microscope, the experimental setup and sample, and information derived by analysis of the images. OME also permits data federation that allows information from multiple sources (e.g., genomic or chemical databases) to be linked to image records. In addition, the OME provides an extensible environment that enables users to write their own applications for image analysis. Consider, for example, the task of tracking labeled vesicles in a time-lapse movie. As noted by Swedlow et al., this problem requires the following: a segmentation algorithm to find the vesicles and to produce a list of centroids, volumes, signal intensities, and so on; a tracker to define trajectories by linking centroids at different time points according to a predetermined set of rules; and a viewer to display the analytic results overlaid on the original movie.2 OME provides a mechanism for linking together various analytical modules by specifying data semantics that enable the output of one module to be accepted as input to another. These semantic data types of OME describe analytic results such as “centroid,” “trajectory,” and “maximum signa,” and allow users, rather than a predefined standard, to define such concepts operationally, including in the machine-readable definition and the processing steps that produce it (e.g., the algorithm and the various parameter settings used).
SOURCE: Based largely on the paper by Swedlow et al. cited in Footnote 145 and on the OME Web page at www.openmicroscopy.org. |
-
Fluorescent speckle microscopy, a technique for quantitatively tracking the movement, assembly, and disassembly of macromolecules in vivo and in vitro, such as those involved in cytoskeleton dynamics;143 and
-
Establishing metrics of similarity between brain images taken at different times.144
These applications are only an infinitesimal fraction of those that are possible. Several research areas associated with increasing the utility of biological images are discussed below. Box 4.7 describes the open microscopy environment, an effort intended to automate image analysis, modeling, and mining of large sets of biological images obtained from optical microscopy.145
As a general rule, biologists need to develop better imaging methods that are applicable across the entire spatial scale of interest, from the subcellular to the organismal. (In this context, “better” means imaging that occurs in real time (or nearly so) with the highest possible spatial and temporal resolution.) These methods will require new technologies (such as the multiphoton microscope) and also new protein and nonprotein reporter molecules that can be expressed or introduced into cells or organisms.
|
143 |
C.M. Waterman-Storer and G. Danuser, “New Directions for Fluorescent Speckle Microscopy,” Current Biology 12(18):R633-R640, 2002. |
|
144 |
M.I. Miller, A. Trouve, and L. Younes, “On the Metrics and Euler-Lagrange Equations of Computational Anatomy,” Annual Review of Biomedical Engineering 4:375-405, 2002, available at http://www.cis.jhu.edu/publications/papers_in_database/EulerLagrangeEqnsCompuAnatomy.pdf. |
|
145 |
J.R. Swedlow, I. Goldberg, E. Brauner, and P.K. Sorger, “Informatics and Quantitative Analysis in Biological Imaging,” Science 300(5616):100-102, 2003. |
The discussion below focuses only on a narrow slice of the very general problem of biological imaging, as a broader discussion would go beyond the scope of this report.
4.4.12.1 Image Rendering146
Images have been central to the study of biological phenomena ever since the invention of the microscope. Today, images can be obtained from many sources, including tomography, MRI, X-rays, and ultrasound. In many instances, biologists are interested in the spatial and geometric properties of components within a biological entity. These properties are often most easily understood when viewed through an interactive visual representation that allows the user to view the entity from different angles and perspectives. Moreover, a single analysis or visualization session is often not sufficient, and processing across many image volumes is often required.
The requirement that a visual representation be interactive places enormous demands on the computational speed of the imaging equipment in use. Today, the data produced by imaging equipment are quickly outpacing the capabilities offered by the image processing and analysis software currently available. For example, the GE EVS-RS9 CT scanner is able to generate image volumes with resolutions in the 20-90 mm range, which results in a dataset size of multiple gigabytes. Datasets of such size require software tools specifically designed for the imaging datasets of today and tomorrow (see Figure 4.5) so that researchers can identify subtle features that can otherwise be missed or misrepresented. Also with increasing dataset resolution comes increasing dataset size, which translates directly to lengthening dataset transfer, processing, and visualization times.
New algorithms that take advantage of state-of-the-art hardware in both relatively inexpensive workstations and multiprocessor supercomputers must be developed and moved into easy-to-access software systems for the clinician and researcher. An example is ray-tracing, a method commonly used in computer graphics that supports highly efficient implementations on multiple processors for interactive visualization. The resulting volume rendition permits direct inspection of internal structures, without a precomputed segmentation or surface extraction step, through the use of multidimensional transfer functions. As seen in the visualizations in Figure 4.6, the resolution of the CT scan allows subtleties such as the definition of the cochlea, the modiolus, the implanted electrode array, and the lead wires that connect the array to a head-mounted connector. The co-linear alignment of the path of the cochlear nerve with the location of the electrode shanks and tips is the necessary visual confirmation of the correct surgical placement of the electrode array.
In both of the studies described in Figure 4.5 and Figure 4.6, determination of three-dimensional structure and configuration played a central role in biological inquiry. Volume visualization created detailed renderings of changes in bone morphology due to a Pax3 mutation in mice, and it provided visual confirmation of the precise location of an electrode array implanted in the feline skull. The scientific utility of volume visualization will benefit from further improvements in its interactivity and flexibility, as well as simultaneous advances in high-resolution image acquisition and the development of volumetric image-processing techniques for better feature extraction and enhancement.
4.4.12.2 Image Segmentation147
An important problem in automated image analysis is image segmentation. Digital images are recorded as a set of pixels in a two- or three-dimensional array. Images that represent natural scenes usually contain different objects, so that, for example, a picture of a park may depict people, trees, and
|
146 |
Section 4.4.12.1 is based on material provided by Chris Johnson, University of Utah. |
|
147 |
Section 4.4.12.2 is adapted from and includes excerpts from National Research Council, Mathematics and Physics of Emerging Biomedical Imaging, National Academy Press, Washington, DC, 1996. |
FIGURE 4.5 Visualizations of mutant (left) and normal (right) mice embryos.
CT values are inspected by maximum intensity projection in (a) and with standard isosurface rendering in (b). Volume rendering (c) using multidimensional opacity functions allows more accurate bone emphasis, depth cueing, and curvature-based transfer functions to enhance bone contours in image space. In this case, Drs. Keller and Capecchi are investigating the birth defects caused by a mutation in the Pax3 gene, which controls musculoskeletal development in mammalian embryos. In their model, they have activated a dominantly acting mutant Pax3 gene and have uncovered two of its effects: (1) abnormal formation of the bones of the thoracolumbar spine and cartilaginous rib cage and (2) cranioschisis, a more drastic effect in which the dermal and skeletal covering of the brain is missing. Imaging of mutant and normal mouse embryos was performed at the University of Utah Small Animal Imaging Facility, producing two 1.2 GB 16-bit volumes of 769 × 689 × 1173 samples, with resolution of 21 × 21 × 21 microns.
SOURCE: Courtesy of Chris Johnson, University of Utah; see also http://www.sci.utah.edu/stories/2004/spr_imaging.html.
FIGURE 4.6 Volume renderings of electrode array implanted in feline skull.
In this example, scanning produced a 131 MB 16-bit volume of 425 × 420 × 385 samples, with resolution of 21 × 21 × 21 microns. Renderings of the volume were generated using a ray-tracing algorithm across multiple processors allowing interactive viewing of this relatively large dataset. The resolution of the scan allows definition of the shanks and tips of the implanted electrode array. Volumetric image processing was used to isolate the electrode array from the surrounding tissue, highlighting the structural relationship between the implant and the bone. There are distinct CT values for air, soft tissue, bone, and the electrode array, enabling the use of a combination of ray tracing and volume rendering to visualize the array in the context of the surrounding structures, specifically the bone surface. The volume is rotated gradually upward in columns (a), (b), and (c), from seeing the side of the cochlea exterior in (a), to looking down the path of the cochlear nerve in (c). From top to bottom, each row uses different rendering styles: (1), summation projections of CT values (green) and gradients (magenta); (2), volume renderings with translucent bone, showing the electrode leads in magenta.
SOURCE: Courtesy of Chris Johnson, University of Utah; see also http://www.sci.utah.edu/stories/2004/spr_imaging.html.
benches. Similarly, a scanned image of a magazine page may contain text and graphics (e.g., a picture of a park). Segmentation refers to the process by which an object (or characteristics of the object) in an image is extracted from image data for purposes of visualization and measurement. (Extraction means that the pixels associated with the object of interest are isolated.) In a biological context, a typical problem in image segmentation might involve extracting different organs in a CT scan of the body. Segmentation research involves the development of automatic, computer-executable rules that can isolate enough of these pixels to produce an acceptably accurate segmentation. Segmentation is a central problem of image analysis because segmentation must be accomplished before many other interesting
problems in image analysis can be solved, including image registration, shape analysis, and volume and area estimation. A specific laboratory example would be the segmentation of spots on two-dimensional electrophoresis gels.
There is no common method or class of methods applicable to even the majority of images. Segmentation is easiest when the objects of interest have intensity or edge characteristics that allow them to be separated from the background and noise, as well as from each other. For example, an MRI image of the human body would be relatively easy to segment for bones: all pixels with intensity below a given threshold would be eliminated, leaving mostly the pixels associated with high-signal-intensity bone.
Generally, edge detection depends on a search for intensity gradients. However, it is difficult to find gradients when, as is usually the case in biomedical images, intensities change only gradually between the structure of interest and the surrounding structure(s) from which it is to be extracted. Continuity and connectivity are important criteria for separating objects from noise and have been exploited quite widely.
A number of different approaches to image segmentation are described in more detail by Pham et al.148
4.4.12.3 Image Registration149
Different modes of imaging instrumentation may be used on the same object because they are sensitive to different object characteristics. For example, an X-ray of an individual will produce different information than a CT scan. For various purposes, and especially for planning surgical and radiation treatment, it can be important for these images to be aligned with each other, that is, for information from different imaging modes to be displayed in the same locations. This process is known as image registration.
There are a variety of techniques for image registration, but in general they can be classified based on the features that are being matched. For example, such features may be external markers that are fixed (e.g., on a patient’s body), internal anatomic markers that are identifiable on all images, the center of gravity for one or more objects in the images, crestlines of objects in the images, or gradients of intensity. Another technique is minimization of the distance between corresponding surface points of a predefined object. Image registration often depends on the identification of similar structures in the images to be registered. In the ideal case, this identification can be performed through an automated segmentation process.
Image registration is well defined for rigid objects but is more complicated for deformable objects or for objects imaged from different angles. When soft tissue deforms (e.g., because a patient is lying on his side rather than on his back), elastic warping is required to transform one dataset into the other. The difficulty lies in defining enough common features in the images to enable specifying appropriate local deformations.
An example of an application in which image registration is important is the Cell-Centered Database (CCDB).150 Launched in 2002, the CCDB contains structural and protein distribution information derived from confocal, multiphoton, and electron microscopy for use by the structural biology and neuroscience communities. In the case of neurological images, most of the imaging data are referenced to a higher level of brain organization by registering their location in the coordinate system of a standard brain atlas. Placing data into an atlas-based coordinate system provides one method by which data taken across scales
|
148 |
D.L. Pham, C. Xu, and J.L. Prince, “Current Methods in Medical Image Segmentation,” Annual Review of Biomedical Engineering 2:315-338, 2000. |
|
149 |
Section 4.4.12.3 is adapted from National Research Council, Mathematics and Physics of Emerging Biomedical Imaging, National Academy Press, Washington, DC, 1996. |
|
150 |
See M.E. Martone, S.T. Peltier, and M.H. Ellisman, “Building Grid Based Resources for Neurosciences,” unpublished paper 2003, National Center for Microscopy and Imaging Research, Department of Neurosciences, University of California, San Diego, San Diego, CA, and http://ccdb.ucsd.edu/CCDB/about.shtml. |
and distributed across multiple resources can be compared reliably. Through the use of atlases and tools for surface warping and image registration, it is possible to express the location of anatomical features or signals in terms of a standardized and quantitative coordinate system, rather by using terms that describe objects in the field of view. The expression of brain data in terms of atlas coordinates also allows it to be transformed spatially to provide alternative views that may offer additional information (e.g., flat maps or additional parcellation schemes). Finally, a standard coordinate system allows the same brain region to be sampled repeatedly to allow data to be accumulated over time.
4.4.12.4 Image Classification
Image classification is the process through which a set of images can be sorted into meaningful categories. Categories can be defined through low-level features such as color mix and texture patterns or through high-level features such as objects depicted. As a rule, low-level features can be computed with little difficulty, and a number of systems have been developed that take advantage of such features.151
However, users are generally much more interested in semantic content that is not easily represented in such low-level features. The easiest method to identify interesting semantic content is simply to annotate an image manually with text, although this process is quite tedious and is unlikely to capture the full range of content in an image. Thus, automated techniques hold considerable interest.
The general problem of automatic identification of such image content has not been solved. One approach described by Huang et al. relies on supervised learning to classify images hierarchically.152 This approach relies on using good low-level features and then performing feature-space reconfiguration using singular value decomposition to reduce noise and dimensionality. A hierarchical classification tree can be generated from training data and subsequently used to sort new images into categories.
A second approach is based on the fact that biological images often contain branching structures. (For example, both muscle and neural tissue contain blood vessels and dendrites that are found in branching structures.) The fractal dimensionality of such structures can then be used as a measure of similarity, and images that contain structures of similar fractal dimension can be grouped into categories.153
4.5 DEVELOPING COMPUTATIONAL TOOLS
The computational tools described above were once gleams in the eye of some researcher. Despite the joy and satisfaction felt when a prototype program supplies the first useful results to its developer, it is a long, long way to converting that program into a genuine product that is general, robust, and useful to others. Indeed, in his classic text The Mythical Man-Month (Addison-Wesley, Reading, MA, 1995), Frederick P. Brooks, Jr., estimates the difference in effort necessary to create a programming systems product from a program as an order of magnitude.
Some of the software engineering considerations necessary to turn a program into a product include the following:
-
Quality. The program, of course, must be as free of defects as possible, not only in the sense of running without faults, but also of precisely implementing the stated algorithm. It must be tested for all
|
151 |
See, for example, M. Flickner, H. Sawhney, W. Niblack, J. Ashley, Q. Huang, B. Dom, M. Gorkani, J. Hafner, D. Lee, D. Petkovic, D. Steele, and P. Yanker, “Query by Image and Video Content: The QBIC System,” IEEE Computer 28(9):23-32, 1995, available at http://wwwqbic.almaden.ibm.com/. |
|
152 |
J. Huang, S.R. Kumar, and R. Zabih, “An Automatic Hierarchical Image Classification Scheme,” ACM Conference on Multimedia, Bristol, England, September 1998. A revised version appears in EURASIP Journal on Applied Signal Processing, 2003, available at http://www.cs.cornell.edu/rdz/Papers/Archive/mm98.pdf. |
|
153 |
D. Cornforth, H. Jelinek, and L. Peich, “Fractop: A Tool for Automated Biological Image Classification,” available at http://csu.edu.au/~dcornfor/Fractop_v7.pdf. |
-
potential inputs, and combinations of factors, and must be robust even in the face of invalid usage. The program should have well-understood and bounded resource demands, including memory, input-output, and processing time.
-
Maintenance. When bugs are discovered, they must be tracked, patched, and provided to users. This often means that the code should be structured for maintainability; for example, Perl, which is extremely powerful, is often written in a way that is incomprehensible to programmers other than the author (and often even to the author). Differences in functionality between versions must be documented carefully.
-
Documentation. If the program is to be usable by others, all of the functionality must be clearly documented, including data file formats, configuration options, output formats, and of course program usage. If the source code of the program is made available (as is often the case with scientific tools), the code must be documented in such a way that users can check the validity of the implementation as well as alter it to meet their needs.
-
User interface. The program must have a user interface, although not necessarily graphical, that is unambiguous and able to access the full range of functions of the program. It should be easy to use, difficult to make mistakes, and clear in its instructions and display of state.
-
System integration and portability. The program must be distributed to users in a convenient way, and be able to run on different platforms and operating systems in a way that does not interfere with existing software or system settings. It should be easily configurable and customizable for particular requirements, and should install easily without access to specialized software, such as nonstandard compilers.
-
General. The program should accept a wide selection of data types, including common formats, units, precisions, ranges, and file sizes. The internal coding interfaces should have precisely defined syntax and semantics, so that users can easily extend the functionality or integrate it into other tools.
Tool developers address these considerations to varying degrees, and users may initially be more tolerant of something that is more program than product if the functionality it confers is essential and unique. Over time, however; such programs will eventually become more product-like because users will not tolerate significant inconvenience.
Finally, there is an issue of development methodology. A proprietary approach to development can be adopted for a number of competitive reasons, ranging from the ultimate desire to reap financial benefit to staying ahead of competing laboratories. Under a proprietary approach, source code for the tools would be kept private, so that potential competitors would be unable to exploit the code easily for their own purposes. (Source code is needed to make changes to a program.) An open approach to development calls for the source code to be publicly available, on the theory that broad community input strengthens the utility of the tools being made available and better enables one team to build on another team’s work.




























































