
4
Challenges of Studying
Environmental Risk Factors
for Breast Cancer
The committee was asked to review the methodologic challenges involved in conducting research on breast cancer and the environment. New insights into carcinogenesis are giving researchers new opportunities to explore both the biology and the epidemiology of breast cancer in relation to environmental exposures. Although progress has been made in investigating the role (whether adverse or not) of environmental factors in breast cancer, the scope of the potential research questions is vast and the questions to be answered are complex. This chapter reviews challenges facing researchers on a variety of fronts, including the nature of the various forms of breast cancer; the diversity and complexity of environmental factors; identifying and measuring exposures at appropriate times; genetic complexity that is still unfolding; and the inherent limitations of the laboratory and epidemiologic tools available to evaluate associations between environmental exposures and disease.
As noted in Chapter 2, breast cancer is a term that captures what is likely to be several diseases. Tumor types have been categorized based on several different characteristics, including age or menopausal status of the woman at the time of diagnosis; the state of the tumor as in situ or invasive; the extent of spread from the initial tumor site; cell type (lobular, ductal); and molecular features of the cells, such as the presence or absence of hormone or growth factor receptors (e.g., estrogen or progesterone receptors [ER or PR], human epidermal growth factor receptor 2 [HER2]). Within
each of these broad categories is considerable variability in tumor characteristics and gene expression. A study examining the gene expression of 65 surgical samples of breast tumors from 42 individuals’ cancers found each tumor to have a distinctive molecular portrait. The tumors showed great variation in their patterns of gene expression, and the variation was multidimensional: different sets of genes showed largely independent patterns of variation (Perou et al., 2000). Further study of the molecular pathology of breast cancer has shed additional light on the possible divergent evolutionary pathways of breast cancer progression, revealing still more complexity (Bombonati and Sgroi, 2011), as discussed in Chapter 5.
While characterizations of tumor and cancer types, such as those noted above, are proving increasingly useful as guides to clinical care and prognosis, their relevance to etiology is not clear. Some associations have been observed between certain tumor types and risk factors (e.g., obesity and ER-positive [ER+]) tumors, but for the most part, the mechanistic basis for these relationships remains to be clarified, as described further in Chapter 5.
Various schematics have been used to illustrate the complexity and interconnectedness of potential factors in breast cancer causation. Howell et al. (2005), for example, illustrate possible roles for genes, pathways, risk factors, modifiable variables, and life events (Figure 4-1). In this representation of some of the known modifiable and unmodifiable risk factors for breast cancer, alcohol serves as an example of a factor that might alter risk for breast cancer in multiple ways. Through induction of aromatase activity, it may foster conversion of androgens to estrogens that have a causal role in breast cancer (Etique et al., 2004). It has also been hypothesized to contribute to genomic instability (Benassi-Evans and Fenech, 2011). Furthermore, it may act indirectly in that its calories can contribute to obesity that itself is associated with breast cancer.
Another illustration (Figure 4-2) of the numerous interrelated factors important in the etiology of breast cancer comes from a complex systems model developed by Robert Hiatt and colleagues as part of a project sponsored by the California Breast Cancer Research Program.1 The developers of this model used expert opinion to select causal factors from four large domains (Societal/Cultural, Physical/Chemical, Behavioral, and Biological) to illustrate the multiple levels of causation that must be considered along with how the factors are integrated across levels and over time. Even though multiple key factors are present, all possible etiologic factors were not included for relative simplicity in interpretation. The model focuses solely on postmenopausal breast cancer because of the different etiologic factors and pathways for premenopausal disease. It takes into account both
![]()
1Personal communication, R. A. Hiatt, University of California, San Francisco, May 21, 2011.
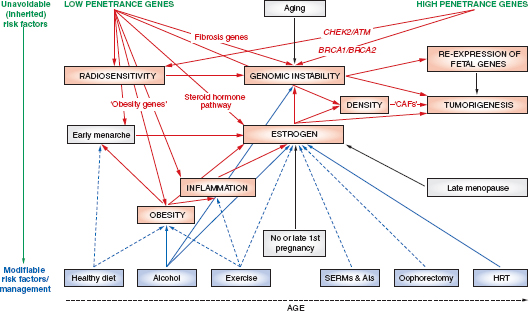
FIGURE 4-1 Overview of risk factors associated with breast cancer. “The diagram summarizes the unavoidable (inherited) and modifiable risk factors that can ultimately lead to tumorigenesis. Genes/pathways/risk factors are shown in red; inherited or unmodifiablefactors are shown in green; modifiable variables are shown in blue; life events are represented by gray boxes; increased/positive effects are denoted by solid arrows; and reduced/negative effects are denoted by dashed arrows. AIs, aromatase inhibitors;ATM, ataxia telangiectasia mutated; BRCA1 and BRCA2 (genes in which deleterious germline mutations increase the risk of cancer); CAFs, cancer-associated fibroblasts; CHEK2, CHK2, checkpoint homolog; HRT, hormone replacement therapy; SERMs, selective estrogen receptor modulators.”
SOURCE: Adapted from Howell et al. (2005, p. 638). Used with permission; Howell, A., A. H. Sims, K. R. Ong, M. N. Harvie, D. G. Evans, and R. B. Clarke. 2005. Mechanisms of disease: Prediction and prevention of breast cancer—cellular and molecular interactions. Nat Clin Pract Oncol 2(12):635–646.
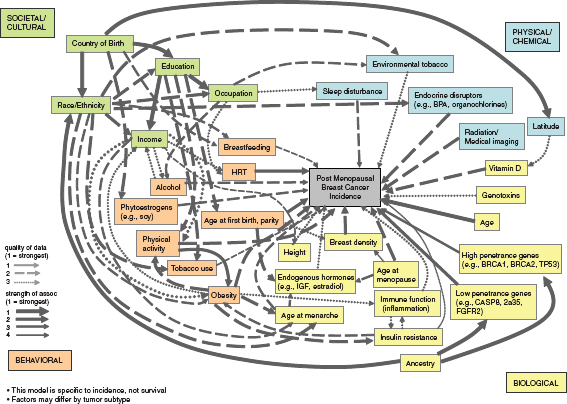
FIGURE 4-2 Illustration of an evidence-based complex-systems model of postmenopausal breast cancer causation. This model displaysmultiple factors associated with postmenopausal breast cancer causation in four broad domains and shows their interconnectionsacross levels(genes to society) by arrows that indicate variations in the strength of the associations and the quality of the data.
SOURCE: Personal communication, R. A. Hiatt, University of California, San Francisco, May 21, 2011. Developed with support from the California Breast Cancer Research Program.
the strength of the associations as well as the quality of the data in the size and hatching of the interconnecting arrows.
Diagrams such as these, which attempt to depict the multiplicity of factors that seem to have a role in breast cancer, help underline the biological complexity of the pathways along which those factors may be acting, the difficulty of distinguishing truly causal effects from associations with intermediate factors, and the challenges of designing, conducting, and interpreting studies that try to evaluate risk factors for the various forms of this disease.
Although these challenges share similarities across the spectrum of risk factors evaluated in this report, they may be particularly acute for evaluating risk relationships from exposures to environmental chemicals. For studies in humans, these include the issues inherent to estimating and assessing exposures, the study design and analytic challenges of environmental epidemiology, and efforts to account for genetic differences in susceptibility to cancer and potential gene–environment interactions. The next portion of this chapter pays particular attention to the challenges in studying environmental chemicals. Studies in animals and in vitro systems pose their own technical obstacles and challenges of interpretation and extrapolation to humans, which are discussed in a subsequent portion of the chapter.
STUDYING ENVIRONMENTAL CHEMICAL AND PHYSICAL EXPOSURES THROUGH HUMAN STUDIES
As noted previously, the committee has adopted a broad approach to the definition of “environment.” A subset of environmental exposures that are of potential concern in the etiology of breast cancer is that of specific chemical and physical agents that might influence breast cancer development. Although information on exposure and the toxicology of many chemicals may be incomplete, for many other chemicals, knowledge of some their properties indicates that they are unlikely to be mutagenic or carcinogenic.
Whether other agents in the environment are able to causally contribute to breast cancer is highly dependent upon both the duration and magnitude (dose) of exposure. One of the most difficult problems in conducting epidemiologic studies on environmental exposures and health effects is to obtain reasonably accurate measurements or estimates of exposures relevant to the disease process. These exposures may occur at low or varying levels or both, for which the relevant time period—the window when the exposure might influence the development of a tumor—is unknown, or they may have occurred years or decades previously. The sections that follow address some of the specific challenges associated with assessing exposures to environmental and physical agents and illustrate the need for additional
or more refined tools to aid in disentangling the possible contributions of these environmental factors to breast cancer.
Assessing Exposures to Chemical and Physical Agents in the Environment
Both the nature of the exposures to chemical and physical agents and the limited means for measuring or assessing them pose challenges for observational research. Human exposures to substances in the environment take place throughout the life course, and in all settings. People are exposed to myriad substances in air, water, and food encountered in homes, schools, workplaces, and even before birth via in utero exposures. A person is exposed not only to individual chemicals, but to mixtures of many different substances, at varying doses simultaneously or at different times. Sometimes it is possible to identify individuals or groups, such as workers in particular occupations, whose typical exposures are considerably higher than those of the average person.
Epidemiologic studies assess whether groups with higher exposures are more likely to experience the outcome of interest, cancer for example, than groups with lower exposures. Determining who is exposed and the degree of their exposures are critical to accurately assessing the association with the health outcome. However, errors in classifying who is more and who is less exposed (exposure misclassification) could limit the ability of a study to show an association with the risk factor where there is one. Thus, accurate exposure assessment is a critical component of human studies to evaluate risk factors for breast cancer or any health outcome.
Historically, studies in occupational settings have been an important means for identifying most chemical carcinogens because in occupational settings, chemical use is often documented and exposure levels tend to be higher than elsewhere. Assessment of exposures in occupational studies are facilitated by extensive sources of data, such as job histories, understanding of production processes and chemicals used, and data from personal or area sampling to measure exposures, as required by the Occupational Safety and Health Administration (OSHA) and standard industrial hygiene practices. Exposure of certain workers to some chemicals may be thousands (or more) times greater than that experienced by the general public, while other workers with different job tasks might experience a wide range of exposures. This variability makes it easier to distinguish people who are exposed to very high levels from those with lesser exposure. The greater the contrast, the firmer the conclusions that can be drawn about differences in risk of disease. When exposure levels are low, contrasts are smaller and exposure misclassification is likely to be relatively greater. Determining exposures can be more difficult in environmental settings, particularly for chemicals that are not regularly monitored in air or food, or for chemicals for which
exposure occurs indoors as a result of specific behaviors or products used. For these reasons, environmental epidemiologic studies are a less effective or efficient approach than occupational epidemiologic studies for demonstrating associations between chemicals and increased rates of disease.
Few of the chemicals identified by the International Agency for Research on Cancer (IARC) or the U.S. Environmental Protection Agency (EPA) as human carcinogens have been classified as such on the basis of studies showing breast cancer in humans. One cannot conclude, however, that these chemicals do not contribute to breast cancer. For virtually all carcinogens identified by IARC and EPA, the evidence base has primarily been from occupational epidemiologic studies for reasons described. For the vast majority of these chemicals, the cohorts were assembled and followed during the 1940s through the 1970s, periods when most industrial firms employed only men.
Historically, therefore, most epidemiologic studies of cancer in the workplace omitted women from the analysis because there were too few present to observe an effect. Because breast cancer is rare in men, such studies lacked the power to detect breast carcinogens. (Power is a function of the expected number of cases of disease in the studies, the level and variability of exposure, the validity of the exposure assessment, and the strength of the true underlying association.) Not only are studies of breast cancer in men underpowered, but also, extrapolation of cancer findings from men to women, which may be justified for other forms of cancer, might not be appropriate for breast cancer.
Beyond the Workplace: Environmental Chemical Exposures
Outside the workplace, exposures to chemicals arise in multiple locations (home, car, ambient air pollution); from multiple activities, including commuting, cleaning, gardening, and smoking; and through different routes of exposure (ingestion, inhalation, dermal absorption).
The home, where people typically spend most of their time2 (Klepeis et al., 1995), provides opportunities for exposure to many chemicals, including naturally occurring chemicals in the diet as well as chemicals from food packaging, processing, or cooking; the release of volatile chemicals from carpets, furniture, clothing treatments, and cleaning products; home use of pesticides; use of cosmetics and personal care products; tobacco smoke; and infiltration of ambient air pollution. Typically, thousands of synthetic and naturally occurring chemicals are present in people’s homes and diet, most at relatively low concentrations.
![]()
2Survey data indicate that on average people spend 69 percent of their time in a residence and 87 percent of their time in enclosed buildings (Klepeis et al., 1995).
The 20th century saw a substantial increase in the synthesis of new chemicals. Tens of thousands of chemicals are used in commerce, and more than 3,000 industrial chemicals (excluding polymers), mostly organic compounds, are produced or imported into the United States at rates exceeding 1 million pounds per year (EPA, 1998b). These are known as high production volume chemicals. A 1998 EPA report found that insufficient testing had been done to evaluate the health effects of all but a few of these chemicals. Of 2,800 chemicals investigated, 93 percent lacked one or more of the six basic toxicity tests,3 and 43 percent of the chemicals had undergone none of these tests, which are considered necessary for a minimum understanding of a chemical’s toxicity. The percentage of chemicals with complete or at least some toxicity information was considerably higher for chemicals with potential for greater exposure through industrial releases or for those in consumer or children’s products. In addition, not all of these 3,000 chemicals are of high priority for testing, because they belong to chemical classes or structural groups for which there is less concern regarding mutagenicity, carcinogencity, or endocrine effects. The High Production Volume Chemicals Program (HPV Program) is an international program to assess the potential hazard of chemicals produced in high volumes. Production levels of specific chemicals can change over time as demand for them increases or declines.
Other chemicals of potential concern are by-products of industrial processes (e.g., dioxins), and the amounts produced cannot be measured as directly as those of deliberately produced chemicals. Opportunities for exposure may change in line with changes in production volumes, but they also may vary independently if industrial processes become more effective in reducing environmental release of a chemical during production. Among the substances reviewed in this report as potential risk factors for breast cancer, environmental releases from different sources have varied, and some have declined over recent years (e.g., dioxin, Figure 4-3 [EPA, 2006]; or perfluorooctanoic acid, Figure 4-4 [Paul et al., 2009]).
Hazard Versus Risk
In the assessment of the impact of environmental chemicals on humans, there is an important distinction between hazard and risk. A chemical may be identified as harmful or a hazard, but the risk it poses to people depends on both its toxic potency and the nature of the exposure, especially the amount to which people are exposed but also potentially the timing of the exposure. While thousands of chemicals are produced in or imported into
![]()
3The tests evaluate acute toxicity, chronic toxicity, developmental and reproductive toxicity, mutagenicity, ecotoxicity, and environmental fate.
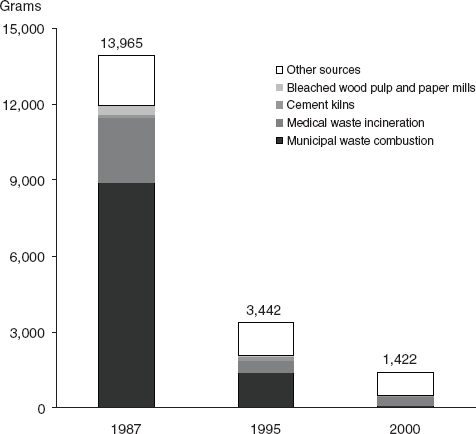
FIGURE 4-3 Sources and amounts (g/yr) of dioxin-like compounds released in the United States in 1987, 1995, and 2000.
SOURCE: EPA (2006).
the United States, not all of them pose risks to the general population. Some are used only in specific industrial processes, where potential exposure is limited to those in the workplace. Some chemicals have low potency, generally causing health effects only at very high exposures. Thus, a chemical known to be a hazard on the basis of toxicologic studies, but with low potency and to which people are exposed at low concentrations, may present little risk of cancer or other adverse health effects.
Route of Exposure
In occupational settings, inhalation and dermal contact are frequently the primary routes of exposure (Eaton and Klaason, 1996), although incidental ingestion pathways can occur. In the home, opportunities exist for
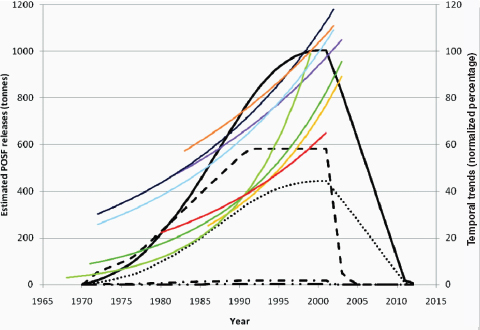
FIGURE 4-4 Estimated releases of perfluorooctane sulfonyl fluoride (POSF) from 1970 to 2012 and exponential temporal trends in biota. POSF breaks down into perfluorooctanesulfonic acid (PFOS). Note: 2012 is when aqueous fire-fighting foams (AFFFs) are scheduled to be restricted and treated carpets end their natural life. The projection to zero is based on 3M’s production only, therefore some emissions will continue from remaining producers. Temporal trends in biota have been normalized to 100 percent for each species/dataset. Usage is depicted as follows: carpets (—), paper and packaging (- • -), apparel (- - -), performance chemicals (– • •), AFFFs (• • •). Biota trend lines are as follows: ringed seals from Arctic locations, Qeqertarsuaq (purple) and Ittoqqortoormiit (yellow); Baltic guillemot eggs (pooled: light green; and mean: dark green); polar bears from western (light blue) and eastern Canadian Arctic (dark blue); herring gulls from Norway (orange); and lake trout from Lake Ontario (red).
SOURCE: Paul et al. (2009, p. 390). Published in: Alexander G. Paul; Kevin C. Jones; Andrew J. Sweetman; Environ Sci Technol 2009, 43, 386–392. Copyright 2008 American Chemical Society.
exposure via ingestion, inhalation, and dermal contact. Pesticide exposures, for example, can occur through consumption of food (from agricultural applications), inhalation (directly from exposure to sprays and foggers or subsequently from volatilization of residues of past use or resuspension of contaminated dust), and dermal absorption (from contact with residues on the surfaces of tables, countertops, or household objects). Various assessments have found that concentrations of some volatile and semivolatile
chemicals are much higher in indoor spaces, such as homes and schools, than in outdoor areas around the home (Sax et al., 2006; Turpin et al., 2007; Ward et al., 2009; Rudel et al., 2010). Dermal exposure may be the predominant exposure pathway for chemicals in some cleaning or personal care products.
Each chemical must be examined for how it is used as well as its volatility and ability to pass through the skin. Sometimes potential routes of exposure can be overlooked—for example, in taking showers, people may experience both dermal and inhalational exposure to some volatile organic compounds (VOCs) in the water supply. Typically, however, this exposure to VOCs is primarily via inhalation and may equal the exposure from drinking water (Jo et al., 1990).
Measurement of Exposure
In occupational studies, job titles and records from industrial hygiene measurements (individual air monitoring, or air sampling from work areas) are frequently used to estimate exposures. For population studies, researchers may use location of residence or distance from a source of concern (transmission wires, freeways, factories); structured questionnaires relying on participants to report product use; measurements taken in air, water, soil, or other environmental media; and measurements in biological specimens (e.g., blood lead, urinary metabolites of pesticides, cotinine from the breakdown of nicotine to indicate tobacco smoke exposure). The utility of these chemical measurements in both environmental and biological samples depends on when the samples are taken relative to the disease in question; the half-life in the environment or human body, respectively; and the variability in actual exposures over time. In the 1990s, researchers began to develop biomarkers as a means not only to improve estimation of exposure, but also to document intermediate steps along the pathway between exposure and effect. For example, markers of oxidative stress, DNA adducts, and epigenetic marks such as methyl groups can provide evidence that tissues have been affected. Such markers may suggest a mechanism by which an exposure may increase or decrease the risk of breast cancer; however, it can be difficult to demonstrate a direct relationship between the exposure and the marker, and between the marker and subsequent disease.
Importance of Timing of Exposure
Understanding the link between chemical exposure and disease is especially challenging when studying chronic diseases that develop gradually over many years, such as cancer. Because the first steps in carcinogenesis may begin decades before the diagnosis of a cancer, relevant exposures
for breast cancer may include those that occurred in childhood or perhaps before birth. Assessing past environmental exposures poses serious challenges.
Chemicals and other factors may act differently or have different exposure routes at different stages of a woman’s life. The breast may be more vulnerable to carcinogenic exposures during in utero development, in the interval between menarche and a first full-term pregnancy, or during key windows of proliferation and maturation. Such periods of increased susceptibility would also imply that total lifetime exposure is not the appropriate metric, but, rather, that exposures need to be measured during critical life stages, some of which may be harder to capture than others. One of the more classic examples of the importance of timing of exposure comes from studies of atomic bomb survivors. Early reports suggested that increased breast cancer risk appeared to be limited primarily to women who were exposed during puberty. Although more recent analyses suggest elevated risks even among those exposed later in life, early exposure remains particularly important (Land et al., 2003; Preston et al., 2007). The potential importance of timing of exposure to breast carcinogenesis is discussed in greater detail in Chapter 5.
Factors for Consideration in Measures of Exposure
A key consideration in epidemiologic studies is persistence of the risk factor or exposure. Some substances are unstable in the environment. Others are retained in environmental media, but have short residence times in the human body. For example, pyrethroid pesticides can persist indoors away from sunlight for months or years, but they are rapidly excreted by humans (CDC, 2009). Some chemicals or their metabolites can be retained in the human body for decades. Levels of internal exposure to these stored chemicals can be influenced by changes in the body that are unrelated to current levels of external exposure. Lead, for example, is stored in bone for decades, but it is released during pregnancy or menopause. Some endocrine-disrupting compounds, such as certain polychlorinated biphenyls (PCBs) and polybrominated diphenyl ethers (PBDEs), are persistent in both the environment and in humans (Brown, 1994; Sudaryanto et al., 2008; EPA, 2010).
If the substance is retained in either environmental media or the human body, it may be possible to make measurements in one time period and infer aspects of exposure in other time periods. To do so requires knowing not only the rate at which a chemical and its metabolites are eliminated from the body (so-called half-lives), but also the variation in exposure levels over time and the determinants of variability in retention (both for the ecosystem and the human organism). Perhaps the most promising situation for infer-
ring previous exposures for the persistent compounds is when they have been banned and no new exposures are occurring, so that half-lives are the prime determinant of change over time. However, even if a typical half-life has been established for a persistent chemical, measurements for individuals can be influenced by such factors as age-related or genetic influences on metabolism of the chemical or experiences that may affect remobilization of the stored chemical (e.g., lactation, substantial change in weight).
If the chemical is not retained, then any one-time measurement will likely be inadequate to capture the true exposure, unless the exposures are consistent. This could occur, for instance, when there is a product whose formulation has not changed and the contact is consistent over time (e.g., used daily in the same amounts and in the same way).
To effectively study exposures over long time periods, research protocols may require measurements of exposure at multiple time points. The number of measurements required will depend on the variation in exposure over time. Compounds that are rapidly excreted may require a large number of measurements, even to obtain estimates of short-term exposures. Because this can be prohibitively burdensome, alternative strategies that rely on external indicators of exposure may provide more accurate estimates of exposure. For instance, if 50 percent of the body burden of a chemical is from consumption of one particular food source, then questionnaires about such behavior patterns may be more reliable than measurements of urinary metabolites.
Thus, new methodologies for the measurement of suspected breast carcinogens in the environment can lead to higher quality epidemiologic studies, both retrospective and prospective. The modalities needed include improving measurements in the environment and assessing variation over time and space; determining routes of exposures and how they vary over time and over the life course; using emissions inventories along with environmental dispersion modeling; measuring compounds and their metabolites in biospecimens; understanding pharmacodynamics and pharmacokinetics and how they vary by age, body weight, nutrition, comorbidity, or other factors; developing biomarkers for early biologic effects (DNA adducts, methylation, tissue changes, gene expression, etc.); using human exposure biomonitoring programs (e.g., breast milk repositories) by geographic areas; and validating exposure assessment questionnaires through various strategies.
Although reductionist science has generally driven typical chemical risk assessments to examine risk “one chemical at a time,” humans are not exposed to just one chemical at a time. Indeed, there is a need to establish new, innovative approaches that allow quantitative assessment of multiple concurrent exposures with various disease end points, such as breast cancer. An interesting conceptual model that addresses the reality of multiple
concurrent exposures is the “exposome.” As proposed by Rappaport (2011, p. 5), the exposome represents “the totality of exposures received by a person during life, encompasses all sources of toxicants and, therefore, offers scientists an agnostic approach for investigating the environmental causes of chronic diseases.” Other examples of recent thinking on the complexities of multiple human exposures include the use of “environment-wide” association studies, such as was done recently for 266 different environmental factors and risk for developing type 2 diabetes (Patel et al., 2010). This study reported small, but statistically significant, associations between development of type 2 diabetes and hepatachlor epoxide, PCBs, and the vitamin gamma-tocopheral. A protective (inverse) effect was seen with beta-carotene. The application of such approaches to the environment and breast cancer is potentially feasible. However, even with such holistic approaches to exposure analysis, assessment of exposure during early life stages would be important, as discussed elsewhere in this report, making it particularly challenging for breast cancer and other diseases that often occur later in life.
Other novel approaches to unraveling the complex association between genes, multiple environmental exposures, and complex diseases such as breast cancer are needed. A better mechanistic understanding of the role of environmental factors in disease etiology, including especially “pathway analyses” and other tools that can be used to identify key regulatory pathways that integrate genetic and environmental modulators, are needed to help set priorities for future research (Gohlke et al., 2009).
HUMAN EPIDEMIOLOGIC STUDY DESIGN AND IMPLEMENTATION
As introduced in Chapter 2, various types of human epidemiologic studies are conducted: (1) double-blind randomized controlled trials, (2) observational designs such as cohort and case–control studies, and (3) the less reliable “ecologic” studies that do not use individual-level data. Each type of study design has different strengths and weaknesses. Several factors are discussed here that can interfere with the execution of studies and interpretation of their findings.
Bias
A major goal of epidemiologic studies is to estimate the effect of a suspected causal factor by measuring how strong its association is with the disease under study. Bias occurs when the estimate of effect systematically misses the mark and is artificially higher or lower than the true association in the population. Bias can arise in several ways. Selection bias occurs when study participants differ from the population of interest with regard to the
joint distribution of exposure and disease. For example, different exclusion criteria applied to cases and controls skewed the prevalences of exposure differently in the two groups in a hospital-based study in Helsinki. This led to an apparent association between the use of reserpine, an antihypertensive agent, and breast cancer (Heinonen et al., 1974). A later study helped to illustrate the false findings resulting from differentially applied selection criteria (Horwitz and Feinstein, 1985; Gordis, 2000).
Information bias can occur when methods for gathering information about study participants are fallible, such that either exposure or disease outcome information is incorrect. Information bias can arise from either random or systematic errors, for example, in information abstracted from medical records or obtained by use of surrogates (e.g., spouses or other family members when the study subject is deceased or too ill to provide information). Recall or reporting bias is thought to be a common type of information bias in case–control studies, but it can occur in any study in which information is obtained after occurrence of disease. One factor that can contribute to recall bias is the tendency of people who develop a disease (cases) to think harder about and recall more potential exposures than those in the comparison group (controls), who have not developed a disease and may be less likely to recollect past events and activities. This type of recall bias would typically result in a bias toward an increased association between the exposures and disease, if those exposures are harmful. However, in general, bias can be either toward the null (the “null” refers to “no effect,” such as an odds ratio [OR] or relative risk [RR] of 1.0) or away from the null. In other words, bias can either attenuate or exaggerate a measure of association.
Another type of bias is referred to as “confounding” and is often considered a category of its own. It occurs when another risk factor for the disease under study occurs more or less frequently in those who are exposed as compared with the unexposed. An association observed between the exposure under study and the disease outcome might be the result of the alternative risk factor that is associated with, but not the result of, the exposure being studied. For example, in the United States, the incidence of breast cancer is generally higher among women with higher incomes, but higher income itself is not a causal factor. Instead, higher income is associated with having fewer children, having children at later ages, and other factors that are more clearly associated with the biologic processes that contribute to breast cancer. For this reason, scientists are skeptical of results unless potential confounders have been taken into account. If confounders are known and measurable, it is often possible to limit the effect of confounding (“control” for it) through appropriate design, data collection, and statistical analysis. For example, matching of controls to cases on factors such as age is a method that can improve the ability to control
confounding when the data are analyzed correctly. But such steps will not be sufficient if confounders are unrecognized or difficult to measure, or if their relationship to exposures is poorly understood.
Restriction of study participants can serve to improve control of confounding, but may result in lower generalizability if the restricted group is characterized by a different relationship between exposure and disease (e.g., a study of women ages 40–60 may yield results that would not apply to women ages 60–80 or 20–40). In matched or unmatched designs, appropriate statistical methods must be applied to effectively control confounding. These include stratification and statistical adjustment. Stratifying the data involves grouping by levels of the suspected confounder and deriving an adjusted measure of association across all levels. Adjustment can involve use of a statistical model that is assumed to reflect the relationship of multiple independent variables in relation to the outcome. Statistical models can provide simultaneous adjustment for multiple potential confounders. However, adjustment for confounders is not always sufficient or adequate, especially when information on all potential confounders has not been collected or the confounding factors are not even known to the investigator.
Moreover, confounders need to be understood as operating, not one-by-one, but rather in a complex network of causal relationships. Graphical tools, such as directed acyclic graphs (DAGs), are sometimes used to identify the appropriate confounders for control, and to identify which factors should not be controlled (Greenland et al., 1999; Hernαn et al., 2002). This latter group consists of two categories of variables: (1) factors that are downstream of the exposure and (2) factors that block a pathway between exposure and disease (e.g., they have antecedents, one that is associated with exposure and the other with disease). Some factors that are downstream of exposure may be intermediates on a causal pathway, but whether they are or not, control for them can introduce bias, except in very specific circumstances (Petersen et al., 2006). In most instances, factors that block an exposure–disease pathway should also not be controlled, in order to obtain unbiased measures of the association of interest. But if they are, at least one of the antecedents will also require control. The importance of taking prior knowledge into account when selecting variables has been clearly demonstrated (Hernαn et al., 2002). Stated differently, the use of empirical data alone for selection of confounders to control can be misleading and can be used to justify what will be a biased model.
Potential confounding by “unknown factors” is often cited as a precaution in ascribing causality to an exposure associated with disease and, in fact, was a central argument used in questioning whether smoking was causing lung cancer (e.g., Fisher, 1958a,b). However, hypothesized explanatory confounders are subject to some stringent constraints, such as that the association between the confounder and disease must be (much)
larger than that of the exposure and disease, in order to “explain” the observed exposure–disease association (Cornfield et al., 1959; Langholz, 2001; Goodman et al., 2002). Consideration of whether such factors exist is warranted when this criticism is expressed. An example was when smoking was argued to explain the association of an occupational exposure with lung cancer. At first blush, this seemed plausible, since smoking has such high relative risks while the occupational exposure showed a lower association. However, smoking levels were found to be very similar when comparing exposed to unexposed workers, and hence smoking was not a strong confounder (Axelson and Sundell, 1978).
Yet another type of bias is statistical bias, which can occur as a result of unavoidable limitations in the methods of analysis. Statistical bias may also result from use of an inappropriate method of analysis, such as using unmatched methods for a matched design, or failing to employ survival analysis when follow-up in a cohort study is highly variable. In addition, a study may be uninformative because it has inadequate statistical power to detect differences in risk when the anticipated effect is small. This may happen because the sample size is too small or, for a rare outcome in a large cohort, the number of expected cases is small or an exposure is rare. Because the study of rare exposures requires such large sample sizes, studies are often conducted in populations that are more highly exposed than the general population, such as occupational groups exposed in the course of their work.
In general, when a study has inadequate statistical power, “random error” could lead to over- or underestimation of the true effect, even though with repeated sampling the average effect estimate would converge on the true value. This imprecision will be reflected in wide confidence intervals around the risk estimate. A wide confidence interval might have an upper limit that is, for instance, 8 or 10 times larger than the lower limit. Another cause of low statistical power is an underlying association that is so small that it is difficult to distinguish from the null effect. For instance, if the exposures encountered by the population truly increase risk by, say, 5 to 10 percent, even a very large study with a few thousand cases will generally not be of sufficient size to reliably generate a statistically significant estimate of increased risk.
Interpretation of Attributable Risk and Population Attributable Risk
Chapter 2 introduced some of the measures that are used to estimate the disease risk associated with factors of interest, including attributable risk (AR) and population attributable risk (PAR). In its simplest form, the AR is a measure (percentage) of the cases that occur in the exposed group that are in excess of those in the comparison group and that are considered
to have occurred because of a given factor. This measure can be interpreted as the maximum potential for risk reduction among those currently exposed to the factor of interest if the exposure could be eliminated and if the association is truly causal. The PAR is a population-based measure of the percentage of excess cases associated with the exposure of interest that also takes into account the distribution of the exposure within the population, again, assuming that the relationship between the exposure and the disease outcome is causal. If an exposure is rare, it may contribute only a small proportion of a population’s disease risk, even if disease incidence is much higher among those who are exposed. The AR is a statement about disease among people who have an exposure, not about exposure among people who have a disease. The PAR is a statement about disease risk ostensibly due to the exposure in the entire population, not about exposure among people with disease.
Several methods have been used to estimate the PAR. A comprehensive review of methodological developments up to 2000 is given in a series of papers, including Benichou (2001), Eide and Heuch (2001), and Uter and Pfahlberg (2001), while more recent developments are described in Steenland and Armstrong (2006) and Eide (2008). With respect to breast cancer, an insightful commentary, “Use and Misuse of Population Attributable Fraction” by Rockhill et al. (1998), succinctly summarizes the definitional and estimation issues and discusses how attributable risk has been misinterpreted in the breast cancer epidemiologic literature.
Although PAR numbers appear fairly frequently in scientific literature, they are prone to misinterpretation by health professionals as well as lay people. It is common to interpret the PAR as the change in disease risk if exposure were reduced in the exposed individuals of the study population. However, exposure may be correlated with other factors that are also risk factors for disease. A change in the exposure under consideration may or may not result in making the “exposure altered” population similar to the unexposed population in all respects relevant to breast cancer. For instance, nulliparous women (those who have never had children) are more likely to be unmarried than parous women. If some of the factors related to being married but not related to childbearing are associated with breast cancer risk, then single nulliparous women who “become parous” to reduce breast cancer risk may not have the same risk as the general parous population, which has more married people in it.
ARs or PARs can be calculated separately for several risk factors related to a disease such as breast cancer, but these separate estimates cannot be added together. It is possible, however, to calculate an AR or PAR for a group of risk factors together. When an AR or PAR is calculated for multiple risk factors combined, the result is likely to be smaller than the sum of the ARs or PARs for the individual factors if the correlations among those
factors are positive. Risk factors are often related to each other or to common disease pathways so their contributions to disease risk are not independent. For example, people who smoke may also consume more alcohol. It is also possible to calculate an AR or PAR for a single factor holding others fixed. The results of some of these studies that have calculated PARs are discussed in Chapter 6.
It is also crucial that ARs and PARs be seen as a function of the characteristics of the source population and mix of exposures from which they are estimated. Because they capture as-yet undetermined factors that contribute to risk, they may not apply to other populations that differ in their mix of risk factors.
Rockhill et al. (1998) point out several common errors in interpretation and communication of the PAR and discuss them in relation to estimated PAR for breast cancer (Seidman et al., 1982; Madigan et al., 1995). The first problem is confusing the attributable risk with the proportion of cases who have any of the risk factors included in the PAR. In the examples they cite, a PAR is reported (say, 25 percent) and then it is erroneously concluded that the PAR proportion (25 percent) of cases have one or more of the risk factors while the remaining cases have no risk factors. As noted, the PAR is a statement about disease risk considered to be due to exposure in the entire population, not about exposure among those with the disease. Rockhill and colleagues noted that in each of these studies, the proportion of controls or of the underlying population who were exposed to at least one of the risk factors was 90 percent or more.
The PAR also does not mean that the causes(s) of breast cancer can be identified for the percentage equivalent to the PAR of those with the disease. The PAR “does not address probability of causation for a specific case of disease, nor does its estimation enable epidemiologists to discriminate between those cases caused by, and not caused by, the risk factors under consideration” (Rockhill et al., 1998, p. 17). A further problem is the lack of distinction between factors that are likely to be causally related to disease risk and those that capture a whole set of lifestyle and exposure characteristics. For instance, they note that denying women a college education (a risk factor for breast cancer) is not going to reduce breast cancer risk if the “more causally proximate exposures and behaviors remain the same” (Rockhill et al., 1998, p. 18).
The third issue has to do with the definition of the exposed group. If the message is that the exposed cases could be prevented, then defining the unexposed group to have characteristics that are unattainable in the exposed population is not useful. In a related note, Rockhill et al. (1998) cite the point by Rose (1985) that susceptibility to chronic disease is rarely confined to a high-risk minority within the population; this would certainly seem to hold for breast cancer.
The last issue is the common practice of “equating the AR with the proportion of disease cases that are ‘explained’ by the risk factors” (Rockhill et al., 1998, p. 18). Their concern is that the term “explained” is equated with, and misinterpreted as, “cause.” A better interpretation might be that the PAR represents the proportion of cases that “are associated with” the risk factors in question. Rockhill and colleagues (1998) note that breast cancer risk factors are poor predictors of breast cancer occurrence; the vast majority of women with these risk factors do not develop breast cancer. As an example to make their point, they consider “age greater than 15” as the exposure variable. Nearly 100 percent of cases are exposed, but so are the vast majority of noncases; therefore, considering age greater than 15 as a risk factor is of little value. This issue can be described in statistical terms as having a defined exposure that has high sensitivity (i.e., a large proportion of breast cancer patients are exposed), but very low specificity (i.e., a large proportion of women without breast cancer are also exposed).
The committee believes that many of the problems in interpretation of the PAR arise when there is either an expressed or implied causal relationship between the exposure and disease. A definition that is more reflective of what may be estimated from observational data is that the PAR is the relative difference in the risk of disease between the whole population and the unexposed portion of the population. With this interpretation, it should be better understood that the lower breast cancer risk in married women is not necessarily due to “marriage” per se, but to some constellation of characteristics of the population of married women.
Experimental Studies in Humans
Many of the various sources of bias and confounding that can affect observational studies are eliminated or reduced in experimental studies or clinical trials that are randomized. In humans, the gold standard for a study to examine a potential causal relationship between an exposure and disease is the randomized controlled trial, in which study participants are randomly assigned to groups receiving (or not) an intervention or exposure. Randomized controlled trials may also be “blinded” when either study subjects or investigators carrying out the study, or both, are unaware of the intervention group assignments. When participants are randomized to receive (or not) a treatment, the likelihood of confounding is reduced, but unless the trial is large, analyses still need to control for the possibility that some imbalance in risk factors (confounding) occurred despite randomization. The randomized trial design is most often used for studies of treatment efficacy, but it is rarely used for etiologic studies.
If large enough, such randomized trials could in theory resolve outstanding questions regarding causal relationships for breast cancer. However,
such studies are not practical or appropriate for most of the environmental exposures of greatest concern. Randomized trials are not ethical if it would be necessary to subject participants to an exposure anticipated by the investigators or the scientific community to be harmful. In many cases, randomly assigning participants to an “unexposed” group would also be infeasible because many substances of interest are widely present in the environment. Nevertheless, randomized trials of intervention strategies to mitigate exposure (e.g., to increase smoking cessation or to reduce worker exposures to a suspected carcinogen) could produce very strong causal evidence if a difference in health outcomes were found.
Another challenge for intervention trials for disease prevention is that assignment to a given intervention may coincide with other changes occurring in the population under study that are not part of the intervention being tested. The Multiple Risk Factor Intervention Trial (the MRFIT study), for example, was designed to test the impact of several interventions on mortality from coronary heart disease (MRFIT Research Group, 1982). After the 7-year study period, investigators found no statistically significant difference in mortality between those who had and had not been part of the intervention group. This unanticipated result was attributed, in part, to those who were assigned to receive “usual care” (the group that did not receive the interventions) experiencing risk-reducing changes (e.g., smoking cessation) that were independent of the study. These studies may also be limited if people who are willing to participate, and to be randomized to the condition of interest, are not particularly representative of the general population at risk. Thus, generalizability of results is often lacking.
Of critical importance for the study of breast cancer are studies that compare women who were “exposed” and “unexposed” during the early life stages for which there is growing concern about higher sensitivities or vulnerability. If such studies were to rely on following women from the time of these exposures, they would have to be carried out over decades to discern differences in rates of breast cancer. A strategy to circumvent this need is for epidemiologists to examine whether the exposure influences an intermediate marker of breast cancer risk, such as age of pubertal onset, that is measureable long before the usual onset of breast cancer.
Interpretation of Group Differences
The interpretation of trends in cancer incidence and mortality in epidemiology requires consideration of multiple simultaneously changing cancer determinants, confounding factors, and even unrelated coincidental trends. Some studies examining statistical associations between health outcomes and exposures or other characteristics make assessments using data at the population or group level rather than the individual level.
Studies that examine population-level associations of disease rates with potential causal factors are termed ecologic studies. They do not look at individuals, but instead look at grouped data for both disease and exposure, such as county rates of cancer (an outcome) and percentages of the county population who have a characteristic (an exposure). Ecologic studies are prone to “ecological fallacy” (Lilienfeld and Lilienfeld, 1980) because sometimes the association seen in the group does not apply to the individuals. For example, counties with high breast cancer rates might also be counties with more women in the workforce, even though within counties, breast cancer might actually occur more often in women who are not employed, or might occur equally in those who are and are not employed. Because of this problem, ecologic studies are considered to be one of the weakest study designs in epidemiology. These designs are best viewed as “hypothesis generating” (a kind of “brainstorming”) rather than “hypothesis testing.”
Impact of Disease Screening
Cancer screening detects asymptomatic cancers. Uptake of screening or dissemination of a more sensitive screening test increases the detection of silent tumors as quickly as the enthusiasm for the new test builds or insurers agree to pay for the test, as occurred in the case of the dissemination of prostate-specific antigen (PSA) screening for prostate cancer. Very few factors other than screening or a sudden shift in diagnostic criteria for cancer can account for rapid changes in cancer incidence. Thus, the implementation of a new screening program or method can account for rapid increases in cancer incidence (Kramer and Croswell, 2009). If the screening process is detecting tumors sooner than they would otherwise have been found, incidence rates are likely to return to previous levels, assuming other factors are not contributing to changes in incidence. New or more extensive screening may also result in a sustained increase in incidence if it detects a reservoir of tumors that routinely exist and would never otherwise have become evident.
Another distinguishing characteristic of screening-mediated increases in cancer incidence compared to appearance of a new carcinogen is the spectrum of tumor stages found at diagnosis. In the absence of screening, the introduction of a new carcinogen would be associated with an increase in the incidence of cancer diagnoses at both localized and advanced stages of cancer. However, screening tests tend to have a disproportionate impact on the incidence of localized stages versus advanced stages because they are finding tumors that cannot otherwise be readily discovered (Kramer and Croswell, 2010).
Screening can also lead to increased detection of indolent cancers that are not life-threatening, a phenomenon known as “overdiagnosis”
(Morrison, 1985). There are two prerequisites for cancer overdiagnosis, and both have been met in the case of breast cancer: (1) the existence of a silent reservoir of tumors that would ordinarily not come to clinical attention during the life span of a given person, and (2) surveillance or screening activities that lead to detection of the reservoir (Esserman et al., 2009; Welch and Black, 2010). Estimates of breast cancer overdiagnosis vary widely (ranging from 7 to 50 percent), depending in part on whether ductal carcinoma in situ (DCIS) is included in the estimate and whether the denominator of the estimate is all cancers or only screen-detected cancers (Gøtzsche and Nielsen, 2006; Zackrisson et al., 2006; Duffy et al., 2008; Gøtzsche et al., 2009). As noted in Chapter 2, at present there is no way to know which instances of DCIS might progress to invasive cancers (Allred, 2010), so most women with in situ tumors receive treatment that is similar to the treatment for early-stage invasive tumors.
Overdiagnosis may also affect the interpretation of study results or surveillance data. Identifying modifiable risk factors that are disproportionately associated with indolent tumors might make it possible to reduce the nominal incidence of breast cancer and spare some women what is essentially unnecessary treatment, but it would have limited benefit for women with more aggressive tumors. Also, because overdiagnosis associated with cancer screening leads to an increase in incidence without necessarily changing the risk of dying of the cancer, it can artifactually inflate survival rates and cure rates of cancer, independent of any actual benefits of screening or improvements in therapy over time (Welch et al., 2000).
Long Latency and Intermediate Markers in Breast Cancer
The process of carcinogenesis usually takes place over many years or even decades. Even the most potent cancer-causing exposure, tobacco smoke, provides an example of long latency in its action. For lung cancer, tobacco smoke is a “complete” carcinogen, meaning that no other exposures are needed beyond smoking to cause cancer. Nevertheless, historically there was a delay of about two decades between widespread uptake of cigarette smoking and the subsequent epidemic of lung cancer, reflecting the latency of the disease.
Because the process of carcinogenesis usually spans years, studying early life exposures that might contribute to or cause cancer is particularly challenging. Of great use would be intermediate outcomes known to be in the causal pathway to cancer, so that studies could use these as endpoints for studying early causes for breast cancer or interventions that could ultimately lower the risk for breast cancer. Currently, candidate intermediate outcomes include early menarche, anovulatory menstrual cycles, greater maximum attained height, late age at first pregnancy, and a small num-
ber of pregnancies. While all are associated with increased risk of breast cancer, the mechanisms or explanations for these associations are not yet established; consequently, it is unknown whether altering the intermediate outcome will also alter the risk for breast cancer later in life. This question may be at the crux of the search for future intervention and prevention strategies.
Moving the Research Agenda Forward
Given the challenges of exposure assessment, timing, and intermediate outcomes outlined above, what are the options for approaching these important questions in human populations using the discipline of epidemiology?
Perhaps an ideal study design would be prospective/longitudinal follow-up of girls from intrauterine life to maturity and past menopause when breast cancer incidence, especially ER+ breast cancer, becomes most common. At frequent intervals over the life course and especially during critical windows of susceptibility, exposure would be assessed by various methods, including self-reports from parents and the girls and women themselves; environmental sampling and measurement; biospecimen assays; and other indicators of impact on physiologic, cellular, or molecular processes. Likewise, assessments of intermediate outcomes that suggest increased risk of breast cancer would also be recorded using the most accurate methods possible.
The obvious problems with this approach are the length of time and the expense needed to capture the data for decades until breast cancers are prevalent in the population. In addition, the type of exposures assessed or targeted along the way may no longer be of interest or relevant a half century later, and the collection, processing, or storage of appropriate specimens may not have been possible at the critical time window. Short of this ideal, what then are the useful, realistic study design alternatives?
No easy answers are apparent. Potentially viable options that could offer useful information are well-conceived monitoring systems and studies addressing intermediate outcomes. For example, large-scale monitoring systems with individual- or group-level information (or a combination of the two) could be leveraged for both prospective and retrospective studies. They could focus on environmental exposures, medical information from electronic health system data, or other sources of relevant exposures, covariates, and intermediate outcomes. Studies could also be designed to systematically focus on (1) relationships between exposures at early phases of development and biomarkers or intermediate outcomes, and linked to other studies of (2) these biomarkers or intermediate outcomes and later risk of breast cancer. Again, leveraging existing databases might lead more rapidly to results.
STUDYING THE ROLE OF GENETICS IN BREAST CANCER
As introduced in Chapter 2, statistical modeling of the potential cumulative effect of the inheritance of multiple risk variants, each of small effect, suggests that low-penetrance gene variants, that is, variants that do not give rise to a strong burden of breast cancer in families, could be associated with a substantial fraction of breast cancer risk. Epidemiologic studies using “candidate gene” approaches have been widely used to assess polymorphic variants in genes that plausibly influence breast cancer risk. More recently, genome-wide association studies (GWAS) have provided a more comprehensive search for associations across the genome, independent of hypotheses about specific genes. GWAS use key variants in single DNA components, called “tag single nucleotide polymorphisms” (tagSNPs), to efficiently evaluate common SNP variations in the human genome (Manolio, 2010). In studies of cases and controls of European ancestry, genotyping of 500,000–600,000 tagSNPs in each study subject permits genome-wide studies of susceptibility to breast cancer. Larger sample sizes are needed for studies of the more variant genomes of persons of African ancestry. As noted in Chapter 2, extreme levels of statistical significance are needed to identify true positive results because of the very large number of statistical tests being performed. Therefore, large sample sizes of thousands, even tens of thousands, of cases and controls are needed for these studies (Hunter et al., 2008).
Thus far, approximately 20 risk variants have been robustly associated with breast cancer risk in GWAS (Easton et al., 2007; Hunter et al., 2007; Stacey et al., 2007; Zheng et al., 2009; Turnbull et al., 2010). A number of these variants are not in regions of genes that code for gene products, and most of the others are not in genes that were previously strong candidates to be associated with breast cancer. Thus, the GWAS approach identifies variation in intergenic regions as potentially important, and it discloses new genes not previously associated with breast cancer, potentially providing new insights into mechanisms of breast cancer causation. Although it is possible that stronger associations may exist for rarer genetic variants (e.g., those with minor allele frequencies of <5 percent) that have not been tested with the technologies available to date, it is unlikely that stronger associations with common variants exist.
Much of breast cancer causation is assumed to be due to the interplay between inherited susceptibility to the disease and exposure to environmental risk factors or lifestyle choices. This interplay is often summarized loosely in the term “gene–environment interaction.” Unfortunately, the
term has several different meanings and mathematical formulations. The generally accepted meaning (Rothman et al., 2008) is that the strength of the association with a given outcome for those with both the high-risk gene polymorphism and the harmful exposure is greater than the sum of the associations for each factor alone. This type of interaction is referred to as “synergistic.” Another use of the term interaction is “statistical,” and this is model dependent. For studies of breast cancer (or any binary outcome measure such as yes/no), the typically used statistical models are all multiplicative, and interaction occurs when the associations for those with both the high-risk gene and the harmful exposure is not multiplicative (e.g., higher or lower). Under this approach, the two factors “interact statistically” if women exposed to both are at much higher risk than would be expected based on multiplying the individual relative risks together.4 Generally speaking, this approach requires a much stronger combined effect than would be necessary to conclude that a synergistic relationship exists. As a result, it can be difficult to replicate findings of statistical interaction.
Because this statistical approach has dominated the breast cancer field, the examples given here test “multiplicative” interaction. The committee notes, however, that biologic interaction can occur through a variety of mechanisms (see Chapter 5), and the synergistic “additive” definition is consistent with factors acting through many of these biologic mechanisms.
Investigating Gene–Environment Interactions
Complex diseases are often the result of both genetic and environmental factors. Few researchers, however, have seriously undertaken the examination of their combined effects. Partly this is because very large studies are needed to identify interactions—for a binary exposure and a gene with two functional forms, the sample sizes need to be at least four times larger than those needed to assess any two-level factor alone with the same statistical power. However, the complexity takes on more dimensions. Even when a study is not investigating the interactions of genes and environmental factors, the ability of those studies to identify environmental risk factors may be compromised by those relationships. Moreover, when exposures become pervasive, all the variability will tend to appear to be due to genetic factors.
For some exposures, investigations of genetic interactions are drawing attention to specific genetic features. The evidence on smoking in conjunction with variants in the N-acetyltransferase 2 (NAT2) gene is discussed
![]()
4This corresponds to the P value for the “multiplicative” interaction term in a logistic regression model. A statistically significant P value with a positive regression coefficient indicates that the joint exposure is associated with higher risk than expected simply by multiplying the relative risks.
in Chapter 3. Some, but not all, studies have suggested that the “slow acetylator” form of NAT2 appears to increase the risk of breast cancer for heavy smokers. Genetic characteristics investigated for interactions with exposure to PCBs and ionizing radiation are discussed here as examples of gene–environment interactions that are being studied.
Polychlorinated Biphenyls
An example of the difficulty of investigating gene–environment interactions is offered by data on polymorphisms in the CYP1A1 gene and exposure to higher blood levels of PCBs. CYP1A1 polymorphisms do not, by themselves, appear to be associated with alterations in risk for breast cancer (Laden et al., 2002; Masson et al., 2005). Similarly, a meta-analysis based on 1,400 case patients with breast cancer and 1,642 control subjects suggests no relation of higher blood levels of PCBs with risk of breast cancer (Laden et al., 2001). Although blood levels of PCBs found in the reviewed studies reflect many years of exposure, the blood samples were mostly collected at the time of breast cancer diagnosis, or less than 10 years before diagnosis, and thus do not exclude an influence of exposures in early life or adolescence.
Other studies have included data on CYP1A1 polymorphisms in the analysis. Moysich et al. (1999) observed in a case–control study with data on 154 postmenopausal cases that women who carried at least one Val allele at codon 462 in the CYP1A1 gene and whose blood levels of total PCB concentration were above the study median had an increased risk of breast cancer (OR = 2.9, 95% CI, 1.2–7.5) compared with women carrying two copies of the Ile allele and below the median for total PCBs (the test for statistical interaction was not statistically significant: P = .13). In a subsequent study based on 293 cases, Laden et al. (2002) reported that postmenopausal women who carried at least one Val allele at codon 462 and were in the highest third of total plasma PCB concentrations had a relative risk for breast cancer of 2.8 (95% CI, 1.0–7.8), compared with women carrying two copies of the Ile allele and in the bottom third of plasma total PCBs (the test for statistical interaction was marginally significant: P = .05). When premenopausal women were included (a combined total of 367 cases), no suggestion of increased risk was evident. In a third study of this association, Li et al. (2005) also observed an increase in risk among women who were above the median value for total plasma PCBs if they were carriers of the codon 462 Val allele (P, interaction .02), although the association was limited to premenopausal cases, not postmenopausal cases.
Thus, these three published studies of this association have reported an elevation in the risk of breast cancer among women jointly exposed to higher plasma PCB levels and the CYP1A1 codon 462 polymorphism,
although the statistical interactions were not all statistically significant, and there was some inconsistency according to menopausal status. Given the possibility that the relevant exposures may have occurred many years before the subjects’ PCB levels were measured, that the particular PCB(s) of concern may not correlate well with the “total PCB” measurements used, and that the small size of the studies limited their power to detect multiplicative interaction, the findings from three different populations are, at a minimum, intriguing and worthy of further investigation.
Ionizing Radiation
Another relation that has been explored is that between mutations in a gene important for DNA repair and exposure to ionizing radiation, which can induce DNA damage. The ATM gene is critical in signaling the occurrence of double-strand breaks and directing repair of the damaged DNA. Furthermore, ataxia–telangiectasia (A-T), an autosomal recessive disorder characterized by extreme sensitivity to radiation, is the result of truncation mutations at the ATM gene. There is strong biological plausibility to the hypothesis that women with ATM mutations, who are less able to respond to DNA damage, will be at higher risk of breast cancer generally and that the breast cancer risk from a given dose of radiation will be greater in women who carry the ATM mutation than in those who do not. In addition, mothers of A-T patients are obligate (heterozygous) carriers of the A-T mutation, and studies indicate that these women are at higher risk of breast cancer than women who do not carry the mutation.
One well-designed study has been done to address the issue of radiation sensitivity. The Women’s Environment, Cancer, and Radiation Epidemiology (WECARE) case–control study draws on women with breast cancer from a consortium of five cancer registries who were followed for a second primary breast cancer (in the contralateral breast). The details about the study design, patient population, and the study results have been reported (Bernstein et al., 2004, 2010; Concannon et al., 2008; Stovall et al., 2008; Langholz et al., 2009). Briefly, blood samples were taken and ATM genotyping performed to locate ATM SNP variants, as well as splicing and truncation mutations, the types of mutations most commonly associated with A-T (Concannon et al., 2008). About 40 percent of women with breast cancer received radiation therapy in the treatment of their disease. The ionizing radiation exposure to the healthy (contralateral) breast could be estimated with a fair degree of accuracy, based on standard practice and treatment records (Stovall et al., 2006). Overall, there was little evidence of variation in risk for a second breast cancer due to radiation across types of ATM variants. However, radiation susceptibility was found in one subset of women whose ATM gene had at least one “rare” variant at SNPs
that both (1) resulted in a change in protein coding (i.e., a “missense” variant) and (2) were highly conserved over species (Bernstein et al., 2010). About 10 percent of breast cancer patients are in this subset, and, in the WECARE Study, the effect of radiation was found to be about twice that observed in women with the same radiation exposure who were not in this subset. Rare variants were defined as those occurring in less than 1 percent of controls, and variation in radiation effect was not seen in more common variants. It is notable that very few cases (15 out of 708) or controls (23 out of 1,397) had the truncation and splicing mutations associated with A-T (Bernstein et al., 2010).
If replicable, these findings suggest that evolutionarily recent changes in the DNA code may impair DNA repair mechanisms, resulting in increased risk of breast cancer due to radiation, but DNA repair is not affected by changes that have “propagated” into the population and become more common. GWAS studies, the current focus of genetic-epidemiologic research, are poorly suited to detect rare SNP changes as they use known marker sites of common genetic variation, and rely on correlation of genetic code locally to detect genetic (or gene–environment interaction) effects. Disease associations with isolated rare SNP changes are not readily detectable by this technique. The WECARE Study results exemplify the difficulty of establishing gene–environment interactions even in the context of an established breast carcinogen and a gene known to be involved in biological mediation of the effect of the carcinogen.
SNP Variants with Robust Associations with Breast Cancer Risk
Another approach to the study of gene–environment interaction is to assess whether the SNP variants robustly associated with breast cancer risk in the GWAS are modified by established environmental or lifestyle risk factors. This approach is not motivated by knowledge of the biological function of specific genes or their relevance to specific exposures. In the largest published report to date, an analysis of 12 such polymorphisms and 10 established risk factors among 7,610 breast cancer cases found no statistically significant interactions after accounting for the 120 interaction comparisons that were made (Travis et al., 2010).
Thus, evidence is limited for robust, replicable “synergistic” interactions between inherited genetic variants of unknown function and established environmental and lifestyle risk factors in breast cancer causation. This by no means makes it irrelevant to quantify these individual associations. For the purposes of risk prediction, all these risk factors appear largely to multiply together—the more genetic or environmental risk factors a women has, the higher her risk. The current evidence merely suggests that the known risk factors do not synergize with the genes that rise to attention
from GWAS approaches, for which the functions are unknown, in a manner that amplifies risk beyond the expectation of multiplying the relative risks for individual factors. Notably, the relative risks associated with the 12 polymorphisms identified by Travis et al. (2010) were small (the maximum was RR = 1.22).
Studies that more systematically address gene–environment interaction are warranted, but the difficulties are considerable. Most published studies have assessed established lifestyle factors, and have limited or no information on hypothesized environmental factors that may be risk factors only in a genetically susceptible subset of women. Very large studies such as the National Institute of Environmental Health Sciences’ Sister Study (www.sisterstudy.org), which has enrolled 50,000 women and collected blood, urine, toenail, and household dust specimens, may provide more information on gene–environment interactions in the future.
Implications of Genetic Variability for Understanding Risk for Breast Cancer
Women who carry a bona fide mutation in BRCA1 or BRCA2 are clearly at such substantially elevated risk of breast cancer that their medical care is altered (Narod and Offit, 2005). Women who carry a higher number of low-penetrance risk alleles are at higher risk than women who carry a lower number (about a 10 percent increase in lifetime risk), but the incremental increase in lifetime risks is far smaller than that of BRCA1/2 carriers compared to noncarriers (estimated to be a 50 percent or more increase in lifetime risk). In an analysis of 5,590 cases and 5,998 controls from the Cancer Genetic Markers of Susceptibility (CGEMS) collaboration, in which the first 10 GWAS-associated SNPs were genotyped, Wacholder et al. (2010a,b) observed that women who carried 13 or more of the 20 risk-conferring variant markers (seen in about 4 percent of the population) had a nearly three-fold increase in risk (OR = 2.90, 95% CI, 2.37–3.55) compared with women carrying six or fewer markers (12 percent of the population). In terms of risk prediction using receiver operating characteristic (ROC) analysis, discrimination between cases and controls on the basis of the number of risk SNPs was relatively poor, but it was equivalent to the discriminatory ability of the clinical standard, the Gail model (Gail et al., 1989), which uses established breast cancer risk factors. Predictions derived from a model that included interactions among the SNPs and the factors used in the Gail model were no better than those from the simpler models. However, women who were classified as at high risk by both the Gail model and the genetic model were at modestly higher risk than women who were at high risk on only one.
In an analysis of 10,306 women with breast cancer and 10,393 con-
trols, in which 7 of the GWAS-associated SNPs were independently confirmed (Reeves et al., 2010), a relative risk of approximately two-fold was observed between the highest quintile of genetic risk and the lowest quintile. The cumulative risk to age 70 for women in the highest of the five genetic risk groups was approximately 7.8 percent, much lower than the 50 to 85 percent cumulative risk associated with BRCA1 or BRCA2 mutations. These authors have previously reported that none of these SNPs were involved in statistical interactions with any established risk factors for breast cancer (Travis et al., 2010).
Thus, when a genetics-based tool for risk estimation becomes available, estimates based on currently established genetic associations of gene variants with breast cancer will contribute to the identification of women at higher risk of breast cancer. However, the relative risks observed are similar to those obtained from risk estimates derived from clinical history and established nongenetic breast cancer risk factors, without the need for genotyping. At present, the combination of genetic and nongenetic risk factors is not deemed to provide sufficient information to enable enough risk stratification to alter the medical care women receive (Reeves et al., 2010; Wacholder et al., 2010a), but this situation may change as more genetic risk loci are discovered.
Another hope for the use of genetic variability in understanding risk is that environmental risk factors that have not been convincingly associated with risk of breast cancer among all women will be convincingly associated with risk among a genetically defined “susceptible” stratum. According to this argument, if only some women are genetically predisposed to breast cancer after exposure to an environmental agent, the environmental signal will not be detected in studies that assess the effect of the environmental factor among all women, whereas if the susceptible women can be identified, the environmental signal will be convincing. At this point, a possible example appears to be that of slow acetylators based on variants in the NAT2 gene and exposure to cigarette smoking. In breast cancer etiology research, most of the assessments have been based on random pairings rather than biologically plausible interactions and have not yielded potentially fruitful leads.
Studies of gene–environment interactions among women with the highest inherited risk, that is, women with pathogenic BRCA1 or BRCA2 mutations, are limited by small sample sizes, and by the fact that women with these mutations identified in the course of clinical care or investigation of familial risk are not systematically enrolled in research studies that include assessment of environmental exposures. Furthermore, prospective studies are limited by many of these women choosing to reduce their future risk of cancer by having bilateral prophylactic mastectomies, removal of their ovaries, or both, once they are identified as BRCA1/2 carriers.
Identifying Important Genetic Factors
As summarized earlier, about 20 genetic loci associated with breast cancer have been discovered since 2007 using the new genome-wide association study approach. Given the sample sizes tested so far, many more loci are likely to be discovered in the near future. Based on the relative risks for SNP variants discovered so far, for a cancer like breast cancer, approximately 50 risk variants with similar characteristics remain to be discovered in larger sample sizes (Park et al., 2010). However, even once these are discovered, much less than half of the inherited variability in breast cancer risk will have been explained (Park et al., 2010). The unexplained variability is sometimes called the “dark matter” of the genome (Manolio et al., 2009). There is a wide variety of opinion on what type of genetic variation may underlie this “missing heritability,” or whether the fraction of breast cancer due to inherited causes may have been overestimated (i.e., more of the true fraction due to inherited susceptibility has been explained than is currently estimated). It remains possible that common and rare variants operating in combination with additional “hits” from environmental factors hold the key to the remaining genetic contributions.
One lesson is clear from the GWAS studies: very large sample sizes of tens of thousands of cases and controls are needed to detect the small to modest relative risks that have typically been associated with common (>5 percent minor allele frequency) variants. This has necessitated the formation of large-scale consortia, such as the Breast Cancer Association Consortium (BCAC), the National Cancer Institute Breast and Prostate Cancer Cohort Consortium (BPC3), and the CGEMS study. Continued enrollment of cases of breast cancer, and appropriate controls, along with collection of DNA, blood, and environmental and lifestyle information, will be necessary to facilitate discovery of more genetic variants, particularly those that confer small additional risk of the disease or act in concert with exogenous (or endogenous) exposures.
Nearly all of the minor allele frequencies of the low-penetrance GWAS-discovered variants are greater than 10 percent. The frequency of disease-associated variants in high-penetrance genes such as BRCA1, BRCA2, p53, and PTEN are much less than 1 percent. The spectrum of genetic variation between 1 and 10 percent is still largely unexplored, and is likely to contribute to an unknown fraction of the “missing heritability” (Figure 4-5). New technologies, notably higher density SNP arrays that allow up to 5 million genotypes to be determined on a single DNA sample, and the advent of sequencing of the whole genome at $1,000 or less, will permit exploration of this genomic territory, but will require large sample sizes to reach statistically robust conclusions.
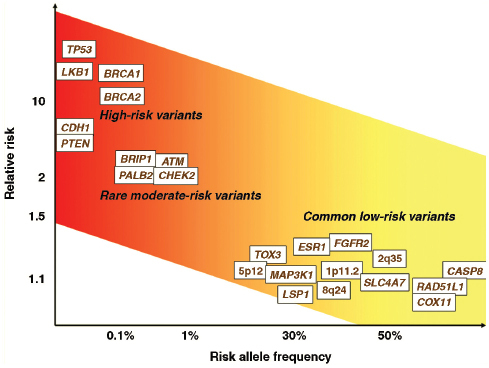
FIGURE 4-5 Genetic variants associated with breast cancer arrayed on the basis of their frequency and their impact on breast cancer risk.
SOURCE: Varghese and Easton (2010). Used with permission: Varghese, J. S., and D. F. Easton. 2010. Genome-wide association studies in common cancers—what have we learnt? Curr Opin Genet Dev 20(3):201–209.
GWAS-style studies of gene–environment interaction require even larger sample sizes, and are limited by the lack of environmental data in many of the case series that have been used for GWAS discovery. A major limitation is the difficulty of establishing exposure to many hypothesized environmental factors, such as exposures that may have occurred in utero, childhood, or early adulthood, or exposures that require sophisticated and potentially expensive biological measurements, especially of biological samples such as adipose or breast tissue. Despite the need for new and improved methods to estimate these exposures, most of the established breast cancer risk factors can be readily ascertained at interview or by questionnaire. Greater emphasis is needed on systematic collection of data on these established risk factors in studies that will be used to generate information on genotypes in order to maximize the sample sizes available for assessment of gene– environment interaction.
STUDYING ENVIRONMENTAL RISK FACTORS THROUGH WHOLE ANIMAL AND IN VITRO EXPERIMENTS
Laboratory animals and human and nonhuman cells and tissue cultures have provided powerful experimental systems for investigating mammary cancer development and mechanisms. As described in Chapter 2, these test systems are also used to identify chemicals that may pose human cancer risks, and provide critical information on how the magnitude and timing of exposure may affect the process of mammary carcinogenesis. This section discusses some challenges in using these model systems, and some emerging approaches that may ultimately provide for improvements in identifying and understanding environmental factors that contribute to breast cancer risk in humans.
In Vivo (Live Animal) Bioassays to Identify Potential Chemical Carcinogens
As briefly outlined in Chapter 2, rodents have long been used to screen chemicals for carcinogenic potential. Standard protocols have been developed that use well-characterized strains of rats and mice and carefully defined approaches for dose selection, length of study, and age windows of exposure. In rodents as in humans, mammary tumors may arise through several modes of action, such as from endocrine-related effects on tissue development and growth or through genotoxic effects on breast cells. Studies in rodent models have also shown the importance of exposure during critical windows of development on mammary cancer risk later in life, in terms of either direct carcinogenic effects or alterations in susceptibility to subsequent exposure to carcinogens. However, the standard protocols for rodent carcinogenicity testing may not be adequate to identify the potential for early-life chemical exposures to induce mammary tumors later in life. Moreover, concerns have been raised about their adequacy for predicting human breast cancer occurrence because of potential differences between rodents and humans in biochemical, cellular, or developmental characteristics (Rudel et al., 2007, 2011; Thayer and Foster, 2007). The following section discusses issues related to these concerns, such as timing of exposure in the bioassay at susceptible age windows, possible confounding by high-dose testing, and differences in species and strain susceptibility.
Timing of Dosing and Windows of Susceptibility
Standard experimental carcinogenicity studies typically begin with young adult animals and thus do not include early life (in utero, perinatal, prepubertal) windows of exposure, which are potentially critical periods
of exposure for some chemicals. The studies often conclude after ongoing exposures for 2 years, or 18 months for some mouse protocols. The average life span of most rodent species and strains used in testing is approximately 2 to 3.5 years. Studies that end well before the completion of the animals’ natural life may miss potential impacts of chemicals that might occur later in life, a period when human breast cancer incidence is high and observed to be modulated, at least by hormone replacement therapy. Rodent bioassays are typically not extended beyond 2 years because of increases in mortality at the maximum tolerated dose and rising numbers of background tumors with age in the controls, both of which compromise statistical comparisons.
Given the existing protocol of beginning chemical dosing after weaning, the most susceptible time for rats to be exposed to mammary carcinogens is for young virgin female rats between the ages of 35 and 60 days (Russo and Russo, 1996; Ren et al., 2008). Rats treated with potent genotoxic carcinogens at 21 days or at over 150 days in age have a much lower frequency of hormone-dependent tumors than rats exposed at 50 days (Medina, 2007). A full-term pregnancy (or administration of pregnancy-related hormones) in rats either shortly before or after exposure to a chemical carcinogen confers considerable protection against development of mammary tumors (Blakely et al., 2006). Parity induces changes in gene expression that are highly conserved among rat strains, thereby conferring increased resistance to development of mammary tumors, even in susceptible rat strains (Blakely et al., 2006). Parity before chemical exposure in mice, likewise, increases resistance to mammary tumor formation (Medina, 2007). In this way, these rodent models are consistent with the observation in humans that a first pregnancy at a younger age and multiple full-term pregnancies are associated with a reduction in breast cancer risk.
Like humans, rats have reproductive cycles that decline with age. Young adult female rats experience estrous every 4 to 6 days. When female rats reach middle age, their estrous cycles become irregular and eventually stop (Morrison et al., 2006). This transitional process is conceptually and functionally similar to menopause in women. However, rodent “estropause” and human menopause are not identical. Some strains of acyclic rats, like the Sprague Dawley strain, actually have chronically high estradiol concentrations and thus are in a state of persistent estrus and lose capacity for a luteinizing hormone surge, whereas Fischer 344 rats and humans develop reduced estrogen secretion as they enter reproductive senescence (Chapin et al., 1996). The time point of initiation of reproductive senescence also differs by rat strain. Similar to humans, it begins at 60 to 70 percent of Fischer 344 rat life span, but starts much earlier in the Sprague Dawley rat, at 30 to 40 percent of its life span (Chapin et al., 1996). Keeping such considerations in mind is important when using rats as models for breast carcinogenesis.
To study “postmenopausal” effects, scientists often use the ovariecto-
mized (OVX) rat model, although this model is not used in standard testing protocols for pesticides and industrial chemicals. Using age-appropriate rats at young, middle, and old ages, the ovaries are surgically removed, and a month later the OVX rats are given estradiol, with or without progestins, in physiologically relevant dosages (Yin and Gore, 2006). There are also other important differences between humans and rodents in postmenopausal hormonal changes, further complicating the use of rodents to study the effects of chemicals on postmenopausal aging, including breast cancer (Morrison et al., 2006).
Various other animal protocols are used to study how exposures during different age windows may affect susceptibility and to examine those chemicals that may have effects in a given age window. Puberty is an important exposure period for later susceptibility for human breast carcinogenesis because of the rapid rate of tissue growth and development. For example, a meta-analysis by Henderson et al. (2010) found that studies were very consistent in showing that exposure to X-irradiation during younger ages (<15) was associated with higher relative risks for breast cancer than was exposure of older age groups, and that the risk declined with increasing age at exposure. Similarly, in rats, the period between the ages of 5 and 8 weeks is one of rapid mammary ductal growth and branching. Fifty-six percent of the rats given a single large dose of the potent genotoxic carcinogen dimethylbenz[a]anthracene (DMBA) during this period developed mammary carcinoma, compared to 8 percent given the same dose at ages less than 2 weeks (Meranze et al., 1969). At this particular point in mammary gland development in the rat, there is a high mitotic index in the terminal ductal structures (Jenkins et al., 2009; Betancourt et al., 2010; La Merrill et al., 2010).
Indeed, administration of DMBA, N-methyl-N-nitrosourea (MNU), or other potent mammary carcinogens on postnatal day 50 is the basis for a widely used experimental rat model of mammary carcinogenicity (Russo and Russo, 1996). This model has also been used to explore the potential for an exposure to an agent or factors much earlier in life to protect against or contribute to mammary tumorigenesis (e.g., olive oil [Pereira et al., 2009]; increased birth weight [de Assis et al., 2006]; and ethyl alcohol [Hilakivi-Clarke et al., 2004]; also see Rudel et al., 2011). Specifically, a number of studies have used models that give a high single dose of a potent genotoxic carcinogen such as DMBA or MNU on or about postnatal day 50 to investigate the effect on mammary carcinogenesis of in utero or perinatal exposures of rats and mice to endocrine-active chemicals (e.g., dioxin plus high-fat diet or BPA) (Jenkins et al., 2009; La Merrill et al., 2010).
Several studies have also used this model to evaluate whether in utero or perinatal exposure to various components of the diet can alter (enhance or inhibit) mammary carcinogenesis. For example, early dietary exposure
to genistein, a soy phytoestrogen (Fritz et al., 1998; Hilakivi-Clarke et al., 1999b), and zearalenone, an estrogenic mycotoxin that frequently contaminates cereal grains (Hilakivi-Clarke et al., 1999b), were observed to be protective; whereas exposure to flaxseed enhanced carcinogenesis (Khan et al., 2007). However, in another study with different exposure characteristics (Hilakivi-Clarke et al., 1999a), genistein enhanced DMBA-induced mammary carcinogenesis. The authors concluded that the direction of effects of genistein on mammary carcinogenesis in the DMBA model was apparently dependent on the dose, length of exposure, and timing.
Ultimately this study protocol may be helpful in identifying some candidate risk factors that may not directly cause tumors later in life themselves, but that may, when exposures occur early in life, modulate risk of mammary tumors associated with subsequent exposure to potent carcinogens. It also raises questions about the predictive value of the standard protocols used in pesticide, pharmaceutical, and industrial chemical testing, as the standard 2-year chronic bioassay protocols (e.g., FDA, 1997; EPA, 1998a; Makris, 2011) generally do not include exposures during in utero and prepubertal periods. Furthermore, standard approaches use exposure to only a single test agent at a time, and thus are not able to identify possible interactive effects with other chemicals, including potential promotional influences on chemically initiated mammary tumors. The animal model of in utero administration of a chemical followed by administration of a potent genotoxic carcinogen at day 50 potentially reflects the ability of early-life exposures to influence the carcinogenicity of other chemicals. However, young children are unlikely to experience a single high dose of a genotoxic substance except in a therapeutic circumstance, such as treatment of cancer with genotoxic chemotherapeutic agents or ionizing radiation. Nevertheless, compared to standard protocols, this model provides information on the potential for cocarcinogenesis in breast cancer—having one exposure that does not appear to alter cancer incidence itself increase the rate of cancer associated with a subsequent exposure.
The National Toxicology Program (NTP), with its emphasis on using 2-year rodent bioassays to identify chemicals that may increase cancer risk in humans, has recently proposed to address the “age at exposure” issue by extending the exposure period for rats to include both in utero and prepubertal exposures (Bucher, 2010). An NTP workgroup on hormonally induced reproductive tumors recommended changes to the standard protocol to include various chemical exposure periods that may be relevant for breast cancer (Thayer and Foster, 2007). NTP indicated in 2010 that it has begun testing some chemicals using its “perinatal protocol”: exposure of the dams begins on gestation day 6 and continues through to postnatal day 21, when the pups are weaned and begin to receive the test compound directly (Bucher, 2010; NTP, 2010c; Foster, 2011).
Species Concordance of Sites of Carcinogen-Induced Tumors
When an excess of tumors is identified in an animal study, questions arise regarding the extent to which such tumors predict human cancer risk from the test agent. While species concordance in overall positive versus negative evidence for carcinogenicity (regardless of organ) is relatively high in standard cancer bioassays (Huff et al., 1991), differences in the specific target site affected are common. In NTP testing reports reviewed by the committee, 90 percent of 30 chemicals that were tested in both mice and rats and that showed mammary carcinogenesis in either species had clear evidence of carcinogenicity (but not necessarily in the mammary gland) in both species (based on cancer occurrence at one or more specific sites). Mammary tumors occurred in both rats and mice for only 17 percent of these 30 tested chemicals, although tumors were induced at other sites. In a committee compilation of information from IARC and NTP reports, 89 percent of agents that IARC has found to have sufficient or limited evidence of human breast cancer also showed evidence of mammary tumors in rats or mice.
Strain and Species Similarities and Differences in Mammary Tumor Susceptibility
Another consideration in the selection of animals for testing is whether the strain used is sufficiently sensitive to detect the carcinogenic activity of an agent that poses a risk to humans. Large differences in species and strain susceptibility for mammary tumors are apparent among rats and mice. Some rat strains are sensitive, while others are resistant to mammary tumors formed spontaneously or induced by hormonal or other agents (Kacew et al., 1995; Ullrich et al., 1996; Thayer and Foster, 2007). For example, in the 1950s diethylstilbestrol (DES) was found to induce mammary tumors in F344 and ACI rats, but the Copenhagen strain was resistant, as was the Sprague Dawley strain in initial studies (Kacew and Festing, 1996). After the link between DES and human breast cancer became clear (IARC, 1987), further testing revealed additional strain differences in sensitivity. Table 4-1 shows some rat and mouse strains that have exhibited sensitivity or resistance to mammary carcinogenesis in research or carcinogenesis screening when exposed to chemical carcinogens (e.g., DMBA, MNU, radiation), hormonal influences, or for mice, the mouse mammary tumor virus (MMTV). The assignment of the relative sensitivity of a strain is somewhat dependent on the agent, its mechanism of action, and tumor type. Rodent strain differences in mammary tumor susceptibility can arise from differences in sexual development, endocrine function, tissue metabolism, or other factors (Kacew et al., 1995; Kacew and Festing, 1996; Bennett and Davis, 2002; Ren et al., 2008).
|
|
||||
| Animal Model | More Sensitive | Less Sensitive | ||
|
|
||||
| Rat | Sprague Dawley Lewis F344/N (fibroadenoma) Wistar-Firth |
Copenhagen Wistar-Kyoto Long Evans F344/N (carcinoma) |
||
| Mouse | BALB/c B6C3F1b C3H RIII |
B6CF1a C57BL/6 O20 |
||
|
|
||||
NOTE: The carcinogenic agents may include chemicals (e.g., dimethylbenz[a]anthracene [DMBA]), radiation, or hormonal influences. For mice, the agent can be the mouse mammary turmor virus (MMTV).
aCross between C57BL/6J and BALB/c.
bCross between C57BL/6J and C3Hf.
SOURCES: Gillette (1976); Kacew et al. (1995); Kacew and Festing (1996); Russo and Russo (1996); Ullrich et al. (1996); Bennett and Davis (2002); Blakely et al. (2006); Boorman and Everitt (2006); Medina (2007); Thayer and Foster (2007); Ren et al. (2008).
Some rat strains show consistency in being highly susceptible (e.g., Sprague Dawley) or resistant (e.g., Copenhagen, Wistar-Kyoto) to most chemical carcinogens, radiation, and hormonal agents, as well as having a corresponding high or low rate of spontaneous mammary tumors (Kacew et al., 1995; Russo and Russo, 1996; Ullrich et al., 1996; Ren et al., 2008). Other strains demonstrate more complex susceptibilities. Fischer 344 rats are susceptible to radiation, some chemical carcinogens such as MNU (although less than Sprague Dawley), and hormonal agents, and they have a high incidence of spontaneous fibroadenomas that increases with age. However, they are also resistant to other chemicals such as N-hydroxy-acetyl-aminofluorene and atrazine (Kacew et al., 1995; Russo and Russo, 1996; Blakely et al., 2006). Long Evans rats are resistant to most chemical carcinogens, but are susceptible (although less so than Sprague Dawley and Lewis) to radiation-induced mammary tumors (Russo and Russo, 1996). Among mice, the B6C3F1 strain is susceptible to the MMTV, radiation, and hormonal agents (Ullrich et al.,1996), although it tends to develop liver rather than mammary tumors from chemical exposure, which may make it less sensitive than the BALB/c strain for testing potential mammary carcinogens (Bennett and Davis, 2002). The BALB/c strain is more susceptible to mammary tumors from chemical and radiation exposure, but it is less sensitive to MMTV (Ullrich et al., 1996; Bennett and Davis, 2002).
Background Tumor Rates and Types
The incidence of specific tumor types in the unexposed animal (the background rate) is another consideration in judging sensitivity of the test animal. Rodent strains used in testing differ in background rates of tumors. High background rates, particularly those that are variable across experiments, may reflect an inherent susceptibility of the animal to exogenous agents and therefore increase the sensitivity of testing for carcinogenicity. Although use of genetically sensitive strains increases the possibility of “false positives” when extrapolating the results to other species, including humans. On the other hand, from a statistical point of view, high background rates can render the study less sensitive if fewer animals are at risk from the test chemical, and more need to develop tumors for the result to achieve statistical significance. Highly variable background rates could also result in the observation of a chemical-related effect by chance, or, conversely, missing a positive response because of an abnormally high background rate in the concurrent control animals. Such considerations make the interpretation of both “positive” and “negative” results challenging in terms of using rodent models to predict human cancer risk.
Table 4-2 shows the background incidences for different types of mammary tumors in strains used in the National Toxicology Program testing program (NTP, 2008, 2010a,b), and compares them to human rates. Overall, the spontaneous lifetime incidence of malignant mammary tumors in female mice and rats was at or below the 12.1 percent lifetime probability of a woman in the United States being diagnosed with breast cancer (NCI, 2010). The low rates in male F344/N rats of 0.16 percent and B6C3F1 mice of 0.09 percent were comparable to the lifetime probability of U.S. men being diagnosed with breast cancer, 0.14 percent (NCI, 2010). Female mice and rats have higher spontaneous lifetime incidence values than males, as occurs in humans.
Benign adenomas occur with a much lower spontaneous incidence in male animals, with rates falling below those for carcinomas for both rat strains and the mouse strain (Table 4-2). Adenomas are tumors of epithelial origin (Donegan, 2002). In women, tubular adenomas have been reported to account for 0.3 to 1.7 percent of benign lesions (Bellocq and Magro, 2003). During pregnancy, tubular adenomas may show secretory changes and are designated as “lactating adenomas.” Lactating and tubular adenomas can be observed in invasive cancer and cancer in situ, although this is uncommon (Bellocq and Magro, 2003). In rodents, tubular adenomas are part of a progression that can be influenced by carcinogen exposure and give rise to adenocarcinoma (Russo and Russo, 1996).
Fibroadenoma is the predominant mammary lesion in certain rat strains, including some used in carcinogenicity screening (Table 4-2). The
| Incidence by Tumor Type (%) | ||||
| Malignant | Benign | |||
| Carcinoma | Adenoma | Fibroadenoma | Fibroma, adenoma, or fibroadenoma | |
| Female | ||||
| Human | 12.15 | Uncommon | 9–28 | 9–28 or more |
| B6C3F1 mouse (n = 1,298) | 1.77 | 0.08 | 0.08 | 0.15 |
| F344/N rat (n = 1,250) | 5.20 | 2.08 | 52.40 | 53.60 |
| Sprague Dawley rat (n = 473) | 10.15 | 2.54 | 67.44 | 68.29 |
| Male | ||||
| Humana | 0.1a | Very rare | Very rare | Very rare |
| B6C3F1 mouse (n = 1,250) | 0.08 | 0 | 0 | 0 |
| F344/N rat (n = 1,298) | 0.39 | 0.31 | 3.16 | 3.62 |
| Sprague Dawley rat (n = 50) | 0 | 0 | 0 | 0 |
aThe majority of male breast cancers are ductal carcinoma; of these, 80 percent of cases are invasive and 2–17 percent are in situ. SOURCES: Komenaka et al. (2006); NTP (2008, 2010a,b); NCI (2010).
importance of these lesions for prediction of human cancer risk is controversial. Certain rat strains used in testing have a very high background rate for them, whereas this tumor type is less common in women. In addition, rat fibroadenoma infrequently progresses to malignancy (Boorman and Everitt, 2006). In the untreated female rats commonly used in NTP background fibroadenoma rates now fall above 50 percent (Table 4-2).
Increased obesity in test animals has contributed to a rise in background incidence of these tumors (Haseman et al., 1998). Harlan Sprague Dawley rats have a significantly higher incidence of mammary gland fibroadenoma (67.4 percent versus 48.4 percent) than the previously used Fisher 344/N strain (Dinse et al., 2010).
Fibroadenomas in women are a benign, biphasic tumor with an epithelial component and a predominant stromal component (Bellocq and Magro, 2003). They are the most common breast tumors in women under age 40, with highest incidence between ages 15 and 35 (Guray and Sahin, 2006), and low incidence in postmenopausal women (Goehring and Morabia, 1997; Kuiper, 2005). In autopsy studies, fibroadenomas were found in from 9 to 28 percent of women (Dixon, 1991; Goehring and Morabia, 1997). Other estimates are that they may be present in 25 percent of asymptomatic women (El-Wakeel and Umpleby, 2003). These estimates suggest that the rates in women are similar to those in earlier NTP rat studies (28 percent). In women, medically important multiple and large fibroadenomas in the breast are seen following treatment with the immunosuppressive drug cyclosporine following organ transplantation (Perera et al., 1986). Other modifiable risk factors are not apparent, and obesity was identified in one review as protective in women (Goehring and Morabia, 1997). Fibroadenomas also occur in men, but they are rare (Rosen, 2001), and they often coexist with gynecomastia (Shin and Rosen, 2007). Cases of fibroadenoma have been reported to follow hormonal treatment for prostate cancer (Shin and Rosen, 2007; Noolkar et al., 2010) and in people who undergo male-to-female sex conversion (Kanhai et al., 1999).
In rats, malignancies will occasionally arise in a fibroadenoma (Boorman and Everitt, 2006). In one series, carcinomas were reported as arising from fibroadenomas at varying low rates in different rat strains (0.5 percent in BN/BiRij, 2.1 percent in Sprague Dawley, 2.3 percent in WAG/Rij) (Huff et al., 1989). In humans, in situ lobular and ductal carcinomas occasionally develop within fibroadenomas (Bellocq and Magro, 2003). There is not agreement as to whether human fibroadenomas can progress to malignancy, and research continues to explore the extent of their relationship to phyllodes tumors, some of which are malignant, and various carcinomas (Markopoulos et al., 2004; Kuiper, 2005; Kabat et al., 2010). Some consider the finding of carcinoma within fibroadenoma as a chance occurrence, and as commonly arising out of similar cells elsewhere in the breast (Dixon, 1991), and that ultimately a fibroadenoma is not a premalignant lesion (Thayer and Foster, 2007). Carcinomas are rarely found within a juvenile fibroadenoma (Rosen, 2009); they are more common in the complex fibroadenoma subtype. Complex fibroadenomas are treated by others as a low probability but a possible source of malignancy (Kuiper, 2005).
High-Dose Testing
The high doses used in carcinogenicity bioassays may complicate interpretation of findings of chemically induced mammary gland tumors. Debates around animal testing often note the potential for tumor induction related to cell proliferation secondary to high-dose cytotoxicity, and that this may be exacerbated by high-dose saturation of metabolic detoxification pathways (Rudel et al., 2007). However, this potential mechanism may be less likely for mammary tumor formation, given the limited fraction of chemicals positive for mammary carcinogenesis in tests conducted with maximum tolerated dosing. Also, unlike the lungs, forestomach, or skin, the mammary gland is not subject to point-of-entry exposure to a chemical. Unlike the liver, the mammary gland also does not receive the first pass of absorbed chemicals from the gastrointestinal tract.
Receptor-mediated responses (in which a chemical must first bind with a cellular receptor to produce the initial biochemical effect), especially those involving multiple receptors and feedback, can first increase and then decrease as dose increases or have other complex, non-monotonic dose– response relationships, as occurs for a variety of endpoints with estrogen and xenoestrogens (Sergeev et al., 2001; Welshons et al., 2003; Watson et al., 2007; Kochukov et al., 2009). This leads to the possibility that the compound has a proportionally different activity in the high-dose experiment than it would at much lower doses. For chemicals that may cause cell proliferation by hormonal mechanisms, such activity may be diminished or eliminated at high doses but present at lower doses. Nevertheless, many of the animal studies demonstrating the hormonal action of environmental chemicals and thereby their influence on carcinogenic risk are conducted at relatively high doses compared to those experienced by humans.
For some chemicals, maximum “tolerated” dosing has the potential to have cytotoxic effects that could either exacerbate mammary through enhanced cell replication as part of an adaptive response to tissue loss, or if sufficiently high, could potentially inhibit carcinogenesis by reducing the population of rapidly dividing cells through cytotoxicity. Either way, high-dose testing that results in cytotoxicity in mammary tissue can leave results of a bioassay for carcinogenesis difficult to interpret or to extrapolate to humans. Add to this the possibility that the carcinogenicity occurs only during a narrow window of development, and the interpretation of any shaped dose–response becomes even more complicated.
Toxicity related to maximum tolerated dosing may also reduce mammary tumor incidence because of reduced weight gain or weight loss (Haseman et al., 1998). Even decreased palatability of the diet due to high concentrations of a test chemical (in the absence of toxicity) can result in decreased weight gain, with attendant decrease in the rate of “spontane-
ous” mammary tumors. Over time, the body weights of rats have steadily increased, and spontaneous mammary tumor incidence correspondingly increased (Haseman et al., 1998). In studies with observed dose-dependent body weight decrements, due to caloric restriction protocols or other factors, there are corresponding reductions in mammary tumor incidence. Thus dose-related reductions in body weight may mask chemical-related increases in mammary tumors. Early mortality due to competing causes of death, including other cancers or toxicity, particularly at the highest dose tested, also decreases study power to observe mammary as well as other tumor types (Rudel et al., 2007).
High doses or more direct administration (e.g., injection) may also change the pharmacokinetics and metabolic pathways of chemicals such that there is a greater (or in some cases lower) proportionate amount of proximate carcinogen formed. High doses administered during pregnancy may result in less maternal sequestering of fat-soluble chemicals such as dioxin and higher proportionate delivery to the fetus than would occur at lower doses. Bolus dosing (rather than more continuous dosing in the diet or drinking water) can also affect the distribution and pharmacokinetics of a chemical. For example, a bolus dose administered directly by stomach tube (gavage) each day is not equivalent to the same amount of chemical ingested continuously over a day. Bolus dosing can result in a larger amount of the chemical or metabolites delivered to cellular targets at one time. Toxicity may be greatly increased if metabolic detoxification pathways become saturated, resulting in greater delivery of the chemical to tissues or greater production of more toxic metabolites through alternative pathways. If, on the other hand, metabolism to more toxic metabolites becomes saturated with bolus dosing, a less than proportional increase in toxicity with dose may result. High bolus dosing may be analogous to an industrial accident or some periodic occupational or medical exposures, but it is unlike much lower episodic or continuous environmental exposures, leaving challenges for interpretation for low-dose risk assessment.
Thus high-dose testing can lead to false positives or false negatives. This poses a dilemma for testing because studies performed using low doses comparable to environmental levels and a standard protocol for numbers of animals per dose group would not be sensitive. A study with 50 animals per dose group can at best detect a statistically significant difference in mammary tumor rate between treated and control animals of 10 to 15 percentage points; any smaller difference would not be detectable because of lack of statistical significance. Thus a hypothetical agent in the background that could be a predominant factor in half the mammary cancers and pose, for example, a difference in incidence of say 6 percentage points, would go undetected in such an animal study if tested at environmental levels. Table 4-3 illustrates this point. It shows the outcome of a hypothetical
| Species and Strain | Number (%) of Animals Expected to Develop Tumors per 50-Animal Dose Groupa | Statistical Significance of Outcomeb | |
| Control | Exposed | ||
| B6C3F1 mouse | 1/50 (1.8%) | 4/50 (7.8%) | 18 |
| Fischer 344 rat | 3/50 (5.2%) | 6/50 (11.2%) | .24 |
| Sprague Dawley rat | 5/50 (10.2%) | 8/50 (16.8%) | .27 |
aTheoretical underlying probability of cancer.
bp value, Fisher’s exact test, showing the probability that the outcome of the experiment is due to chance alone. A p value of .05 or less typically indicates statistical significance.
experiment in which the exposed group of animals receives an environmentally relevant dose of an agent that results in an increase in the incidence of tumors (6 percentage points) equivalent to half the average lifetime risk of breast cancer in the United States. The control groups reflect background rates of mammary carcinomas observed in NTP studies (see Table 4-2). The tumor rates in the exposed groups reflect the expected response in the controls, elevated by 6 percentage points—the effect expected from the chemical exposure. For no case would the results in the exposed group be found to be statistically significant, given a sample size of 50 animals and the background tumor rates in the controls.
Species Similarities and Differences in Mammary Gland Biology and Carcinogenesis
Rodents used in carcinogenicity experiments are generally similar to humans in mammary gland development, although some aspects of the timing may differ. For example, the epithelial bud and ductal outgrowth occurs late in gestation for rodents, but the evidence suggests ductal development occurs early in gestation for humans (Fenton, 2006; Table 4-4).
As with humans, female mice and rats have greater occurrence of spontaneous mammary tumors than males, reflecting endocrine-related influences (see Table 4-2). Estrogen plays an important role in breast cancer in rodents and humans. Certain other hormones—such as prolactin, progesterone alone, and androgens—appear to be involved in inducing mammary tumors in rodents, but have a less clear role in the induction of breast cancer in humans (Thayer and Foster, 2007). However, the recent evidence for the role of prolactin and progesterone in human cancer has become stronger (Fernandez et al., 2010; Jacobson et al., 2011).
TABLE 4-4 Timing of Events in Mammary Development in Humans and Rodents
|
|
||||
| Developmental Event | Human | Rodent | ||
|
|
||||
| Milk streak evidence | EW 4–6 | GD 10–11 (mice) | ||
| Mammary epithelial bud forms | EW 10–13 | GD 12–14 (mice) GD 14–16 (rats) |
||
| Female nipple and areola form | EW 12–16 | GD 18 (mice) GD 20 (rats) |
||
| Branching and canalization of epithelium | EW 20–32 | GD 16 to birth (mice) GD 18 to birth (rats) |
||
| Secretion is possible | EW 32–40 (ability lost postnatally) | At birth with hormonal stimuli | ||
| Isometric development of ducts | Birth to puberty | Birth to puberty | ||
| Terminal end buds present (peri-pubertal) | 8- to 13-year-old girls | 23- to 60-day-old rodents | ||
| Formation of lobular units | EW 32–40, or within 1–2 years of first menstrual cycle | Puberty and into adulthood | ||
|
|
||||
NOTES: EW, embryonic week; GD, gestational day.
SOURCE: Fenton (2006, p. S19). Used with permission: Fenton, S. E. 2006. Endocrine-disrupting compounds and mammary gland development: Early exposure and later life consequences. Endocrinology 147(6 Suppl):S18–S24. Copyright 2006, The Endocrine Society.
As also noted above, tumor types in rats and humans are similar (Russo and Russo, 1996; Fenton, 2006), although the background rates of tumors may differ (see Table 4-2). Premalignant changes can also be observed in both rats and humans (Thayer and Foster, 2007). Rats, however, may be poor animal models for studying metastasis in humans because their mammary gland carcinomas rarely metastasize (Thayer and Foster, 2007). By contrast, mouse mammary tumors often metastasize to the lungs (Cardiff and Kenney, 2011). Although spontaneous and virally induced mouse mammary tumors differ in histology and morphology from breast tumors in humans, tumors in genetically engineered mice resemble those in humans (Cardiff and Kenney, 2011).
Certain modes of action hypothesized for the rat have been questioned regarding their relevance to humans, such as mammary tumors that result from atrazine exposure, presumably through lengthening of the estrous cycle and elevation of endogenous levels of prolactin and estradiol in Sprague Dawley but not F344 rats (Kacew et al., 1995). Induction of mammary tumors in mice by viral origin does not appear to be relevant for
humans, but it remains a useful model in studies of expression of oncogenes (Medina, 2010).
Similarities and Differences Between Rodents and Humans in Metabolism of Carcinogens
One of the most common reasons why one species may differ from another in terms of tumor development following exposure to a chemical carcinogen is because of differences in the way the carcinogen is biotransformed (metabolized). More than 95 percent of known chemical carcinogens require biotransformation to reactive intermediates to exert their carcinogenic effects, typically through an enzyme-mediated oxidation reaction. These reactive intermediates are often quickly eliminated through other biotransformation processes, such as conjugation or hydrolysis. Because biotransformation of xenobiotics is in part a function of “adaptive response” to one’s environment, evolutionary influences have resulted in relatively large genetic divergence in xenobiotic biotransformation pathways, giving rise to potentially important species differences in susceptibility to carcinogens. For example, mice express a particular form of glutathione S-transferase (GST) with remarkably high catalytic efficiency toward detoxification of the potent liver carcinogen, aflatoxin B1. Neither rats nor humans express this particular form of GST in the liver, and consequently both species are highly sensitive to the hepatocarcinogenic effects of aflatoxins (Eaton and Gallagher, 1994). Thus, understanding species- and tissue-specific pathways involved in carcinogen activation and detoxification is an important element of “predictive toxicology” that relies on animal models.
Biotransformation of xenobiotics can occur in virtually any tissue, although the majority of “clearance” of a chemical from the body through biotransformation reactions usually occurs in the liver. However, for some carcinogens that act at sites distant from the point of exposure, biotransformation in the target tissue may be critically important. Even if it does not contribute substantially to the overall elimination of the substance from the body, tissue-specific biotransformation could be significant for activating a compound to a carcinogenic form in a particular tissue. Thus, for potential mammary carcinogens, it is important to understand if there are tissue-specific differences in biotransformation of xenobiotics in breast tissue in humans versus experimental animals. In addition, it is important to understand potential differences in the formation and systemic transport of reactive intermediates from the liver (Ioannides, 2002).
For most lipophilic genotoxic carcinogens, activation to reactive intermediates occurs via the cytochrome P-450 (CYP) superfamily of enzymes (Nebert and Dalton, 2006). CYP expression in mammary tissue is poten-
tially important in both activation and detoxification of polycyclic aromatic hydrocarbons (PAHs), aromatic amines, and other genotoxic “procarcinogens.” Furthermore, CYPs may also play a role in the elimination of estrogenic compounds that partition into mammary lipids, thus potentially playing a protective role for estrogen-active compounds at the site of action. They also play a role in steroid hormone biosynthesis, and induction or inhibition of some CYPs can affect the levels of estrogens and other hormones.
There are 57 different genes, in 18 gene families, in the human genome that code for CYP enzymes (Nelson et al., 2004). A subset of these genes and their related enzymes are of particular relevance to breast cancer because they are involved in xenobiotic biotransformation to active or inactive agents or can be modulated by other xenobiotics to affect steroid hormone levels or steroid genotoxicity. These include the CYP1 family (which has three members: CYP1A1, CYP1A2, and CYP1B1), as well as CYP2E1, CYP3A4, and CYP19. For example, the CYP1A1 enzyme is involved in the activation of PAHs to reactive diol-epoxides, and thus is very important in the carcinogenicity of PAHs. PAHs cause mammary tumors in rats (Cavalieri et al., 1988), and they are biotransformed by human mammary epithelial cells to metabolites that form PAH–DNA adducts (Calaf and Russo, 1993).
Whether activation of PAHs to mutagenic metabolites via CYP1A1 occurs directly in breast tissue is not completely clear, although it is likely because CYP1A1 mRNA (Huang et al., 1996) and CYP1A1 are identified in human breast tissue (Hellmold et al., 1998). PAH–DNA adducts have also been identified in normal breast tissue, and were reported to be higher in women with breast cancer than in healthy controls (Li et al., 1999). Thus, it appears that, at least in some individuals, human breast tissue has the capacity to activate PAHs and potentially other procarcinogens to DNA-reactive molecules. CYP1A1 is inducible in lung and oral mucosal tissue by exposure to cigarette smoke, although no published reports demonstrating that smoking induces expression in human breast tissue were found. It is also inducible in mouse and human mammary tissue by a number of other xenobiotics, including 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). The degree to which species differences in tissue-specific expression of xenobiotic biotransformation enzymes might contribute to species differences in response to breast carcinogens evaluated in chronic rodent bioassays is an issue for consideration in selection of animals for carcinogenicity testing.
Nonstandard Whole Animal Carcinogenicity and Related Studies
Mouse mammary tumors and the associated MMTV were among the first tumor types investigated in studies of animal carcinogenicity, including
research on neoplasia, oncogenic viruses, host responses, role of endocrinology and stem cells, and progression. More recently, genetically engineered mice have been used to investigate the role of agents such as the MMTV in promoting Myc oncogene expression in the mammary gland. Genetically engineered mice have allowed considerable research into the molecular biology of mammary tumor formation and progression.
Since the development of the first model of breast cancer in a genetically modified mouse, more than 100 models have been developed to study breast cancer (Cardiff and Kenney, 2011). At least three basic types have been developed based on transgenes, combinations of transgenes, and targeted mutations. Gene targets have included growth factors and their receptors, cell signaling pathways, cell cycle regulators, and differentiation mediators (Bucher, 2010). The advantage of these models is that they have a defined genetic background, enabling the study of particular pathways, without variations attributable to differences in genotype. The mice develop disease after a predictable time period, and the stage-specific alterations directly translate to humans (Bucher, 2010). Finally, they correspond well to humans in that mammary tumors in mice are caused by the genes that are overexpressed or mutated in human breast cancer (Bucher, 2010).
In contrast to most breast cancers in humans, most mouse tumors (including those in many genetically modified mice)—whether of spontaneous, viral, or chemical origin—do not express hormone receptors (Lanari et al., 2009). To aid in the investigation of human breast cancers, however, specific mouse models have been developed in which mammary tumors do express hormone receptors (e.g., estrogen and progesterone receptors) and demonstrate other molecular features found in human tumors (e.g., Lanari et al., 2009; Herschkowitz et al., 2011; Nguyen et al., 2011). For one of these models, medroxyprogesterone acetate (MPA) is administered to BALB/c female mice, resulting in hormone-dependent, metastatic mammary ductal carcinomas that express both estrogen and progesterone receptors, as in humans (Lanari et al., 2009). Another model relies on transplantation of mammary cells without p53 tumor suppressor function into BALB/c mice (Herschkowitz et al., 2011). Models such as these allow investigation into factors affecting hormone dependence of carcinomas, as well as those that promote tumor progression or regression. Further research is needed on the use of genetically modified mouse models for assessing the effect of environmental exposures in inducing tumors.
Some studies are designed to assess the impact of a test compound on mammary development by periodic evaluation of whole mounts of mammary tissue from animals during the postnatal period (Fenton et al., 2002; Birnbaum and Fenton, 2003; Rudel et al., 2011). The test compound is typically given in utero or neonatally, and mammary structures are evaluated against those of control animals to determine treatment-related differ-
FIGURE 4-6 Development of the mammary gland in rats following in utero exposure to atrazine (ATR) and 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD).
SOURCE: Birnbaum and Fenton (2003). Reproduced with permission from Environmental Health Perspectives.
ences in development. In particular, branching density, bud formation off the major ducts, and terminal end bud formation are evaluated. Figure 4-6 shows mammary gland whole mounts from Long Evans rats exposed during late gestation to atrazine or TCDD (Birnbaum and Fenton, 2003). The tissue preparations show the differences in key developmental parameters on different postnatal days (PNDs). Mammary development was observed to be disrupted as early as PND 4. For example, in treated animals, there was a lack of branching, fewer primary ducts from the nipple, and fewer terminal structures. These are examples of how such a test system could be used to evaluate the potential of a chemical to modulate mammary development. The degree to which changes in mammary structure that result from early exposures signal increased sensitivity to mammary tumor development is an area for study to increase the usefulness of these assays for detecting potential breast carcinogens (e.g., Rudel et al., 2011).
In Vitro Studies
In vitro tests, discussed briefly in Chapter 2, have been used as initial, putative predictors of carcinogenicity potential. Genotoxicity has long been
treated as an indicator of possible carcinogenicity. Testing requirements for pesticides and pharmaceuticals include in vivo studies in rodents to test for chromosomal aberrations and micronuclei, and in vitro tests for mutations in mammalian cells and bacteria. Tests based on mechanisms other than genotoxicity are also used. The goal is to target an agent’s ability to modulate pathways that underlie the basic mechanisms of toxicity. While regulatory agencies typically do not label chemicals that have tested positive in genotoxicity tests as possible carcinogens in the absence of supporting human or animal data, product development programs for pesticides and pharmaceuticals often avoid chemicals with strong signals of genotoxic potential, mostly out of concerns for potential carcinogenicity and mutagenicity.
Because the current standard whole-animal testing approach for carcinogenicity (as well as other endpoints) is time- and resource-consuming, initiatives to move toward reliance on in vitro and structure–activity relationships have been advocated and are under way (NRC, 2007; Krewski et al., 2009; EPA, 2011). The National Research Council (NRC) has envisioned a new toxicity testing system, focusing on upstream events: chemical perturbations of cellular response networks (i.e., complex biochemical interactions that maintain normal cellular function) (NRC, 2007). For example, testing might identify perturbation of estrogen signaling and the subsequent events that potentially result in cancer. The NRC vision was made possible because of the emerging scientific understanding of cellular response networks, and high-throughput technology that enables the exploration of the structure of these networks and rapid conduct of in vitro tests. NRC (2007) proposed the development of suites of predictive, high- and medium-throughput assays, emphasizing those based on cells of human origin, to evaluate perturbations. These would be complemented by assays of more integrated cellular responses and in vivo assays to cover uncertainties in the testing regimen, to test prototypic compounds, and to address metabolism. Other components of the framework include the use of physiologically based pharmacokinetic studies, human biomonitoring data, and epidemiologic data to evaluate and fine-tune the predictive ability of the tests. The NRC vision was accompanied by a long-term strategy for its realization, involving a substantial multidisciplinary research program.
Subsequently, in 2008, various federal institutions entered into a Memorandum of Understanding to “research, develop, validate and translate innovative chemical testing methods that characterize toxicity pathways” (EPA, 2011). The current “Tox21 collaboration,” renewed for 5 years in 2010, includes EPA, the NTP, the National Human Genome Research Institute and the Chemical Genomics Center of the National Institutes of Health, and the FDA. The main work will be to explore high-throughput screening assays and tests using phylogenetically lower animal species (e.g.,
fish, worms), and high-throughput whole-genome analytical methods to evaluate mechanisms of toxicity. The ultimate end is to generate data with the new tools for use in the protection of human health and the environment.
An important consideration in the development of tests that have adequate coverage will be the degree to which they cover the pathways involved in the general mechanisms underlying breast cancer—mutagenesis, estrogen receptor signaling, epigenetic programming, growth promotion via mitogenic cell signaling, microenvironmental change, and modulation of immune functioning. This will require attention in selection of cell types and environments relevant to breast cancer.
Better understanding of the contribution of environmental factors to breast cancer entails understanding the multiple challenges in carrying out and interpreting studies in humans, animals, and in vitro systems. For studies in humans, these include the issues inherent in estimating and assessing exposures, the study design and analytic challenges of environmental epidemiology, and efforts to account for genetic differences in susceptibility to cancer and potential gene–environment interactions. Studies in animals and in vitro systems bring with them their own technical obstacles and challenges of interpretation and extrapolation to humans. An understanding of these challenges informs understanding of the existing data and their implications for next steps for action and research.
Allred, D. C. 2010. Ductal carcinoma in situ: Terminology, classification, and natural history. J Natl Cancer Inst Monogr 41:134–138.
Axelson, O., and L. Sundell. 1978. Mining, lung cancer and smoking. Scand J Work Environ Health 4(1):46–52.
Bellocq, J. P., and G. Magro. 2003. Fibroepithelial tumours. In Pathlogy and genetics of tumours of the breast and female genital organs. Edited by F. A. Tavassoli and P. Devilee. Lyon, France: IARC Press.
Benassi-Evans, B., and M. Fenech. 2011. Chronic alcohol exposure induces genome damage measured using the cytokinesis-block micronucleus cytome assay and aneuploidy in human B lymphoblastoid cell lines. Mutagenesis 26(3):421–429.
Benichou, J. 2001. A review of adjusted estimators of attributable risk. Stat Methods Med Res 10(3):195–216.
Bennett, L. M., and B. J. Davis. 2002. Identification of mammary carcinogens in rodent bioassays. Environ Mol Mutagen 39(2–3):150–157.
Bernstein, J. L., B. Langholz, R. W. Haile, L. Bernstein, D. C. Thomas, M. Stovall, K. E. Malone, C. F. Lynch, et al. 2004. Study design: Evaluating gene–environment interactions in the etiology of breast cancer—the WECARE study. Breast Cancer Res 6(3):R199–R214.
Bernstein, J. L., R. W. Haile, M. Stovall, J. D. Boice, Jr., R. E. Shore, B. Langholz, D. C. Thomas, L. Bernstein, et al. 2010. Radiation exposure, the ATM Gene, and contralateral breast cancer in the Women’s Environmental Cancer and Radiation Epidemiology Study. J Natl Cancer Inst 102(7):475–483.
Betancourt, A. M., I. A. Eltoum, R. A. Desmond, J. Russo, and C. A. Lamartiniere. 2010. In utero exposure to bisphenol A shifts the window of susceptibility for mammary carcinogenesis in the rat. Environ Health Perspect 118(11):1614–1619.
Birnbaum, L. S., and S. E. Fenton. 2003. Cancer and developmental exposure to endocrine disruptors. Environ Health Perspect 111(4):389–394.
Blakely, C. M., A. J. Stoddard, G. K. Belka, K. D. Dugan, K. L. Notarfrancesco, S. E. Moody, C. M. D’Cruz, and L. A. Chodosh. 2006. Hormone-induced protection against mammary tumorigenesis is conserved in multiple rat strains and identifies a core gene expression signature induced by pregnancy. Cancer Res 66(12):6421–6431.
Bombonati, A., and D. C. Sgroi. 2011. The molecular pathology of breast cancer progression. J Pathol 223(2):307–317.
Boorman, G., and J. I. Everitt. 2006. Neoplastic disease. In The laboratory rat, 2nd ed. Edited by S. M. Suckow, S. H. Weisbroth, and C. L. Franklin. American College of Laboratory Animal Medicine Series. Boston, MA: Elsevier Academic Press.
Brown, J. F. 1994. Determination of PCB metabolic, excretion, and accumulation rates for use as indicators of biological response and relative risk. Environ Sci Technol 28(13):2295–2305.
Bucher, J. 2010. Weighing evidence from National Toxicology Program cancer bioassays. Presentation at the Institute of Medicine Breast Cancer and the Environment meeting, July 6–7, San Francisco, CA.
Calaf, G., and J. Russo. 1993. Transformation of human breast epithelial cells by chemical carcinogens. Carcinogenesis 14(3):483–492.
Cardiff, R. D., and N. Kenney. 2011. A compendium of the mouse mammary tumor biologist: From the initial observations in the house mouse to the development of genetically engineered mice. Cold Spring Harb Perspect Biol 3(6).
Cavalieri, E., E. Rogan, and D. Sinha. 1988. Carcinogenicity of aromatic hydrocarbons directly applied to rat mammary gland. J Cancer Res Clin Oncol 114(1):3–9.
CDC (Centers for Disease Control and Prevention). 2009. Fourth national report on human exposure to environmental chemicals, 2009. Atlanta, GA: CDC. http://www.cdc.gov/exposurereport/index.html (accessed October 16, 2011).
Chapin, R. E., J. T. Stevens, C. L. Hughes, W. R. Kelce, R. A. Hess, and G. P. Daston. 1996. Endocrine modulation of reproduction. Fundam Appl Toxicol 29(1):1–17.
Concannon, P., R. W. Haile, A. L. Borresen-Dale, B. S. Rosenstein, R. A. Gatti, S. N. Teraoka, T. A. Diep, L. Jansen, et al. 2008. Variants in the ATM gene associated with a reduced risk of contralateral breast cancer. Cancer Res 68(16):6486–6491.
Cornfield, J., W. Haenszel, E. C. Hammond, A. M. Lilienfeld, M. B. Shimkin, and E. L. Wynder. 1959. Smoking and lung cancer: Recent evidence and a discussion of some questions. J Natl Cancer Inst 22(1):173–203.
de Assis, S., G. Khan, and L. Hilakivi-Clarke. 2006. High birth weight increases mammary tumorigenesis in rats. Int J Cancer 119(7):1537–1546.
Dinse, G. E., S. D. Peddada, S. F. Harris, and S. A. Elmore. 2010. Comparison of NTP historical control tumor incidence rates in female Harlan Sprague Dawley and Fischer 344/N Rats. Toxicol Pathol 38(5):765–775.
Dixon, J. M. 1991. Cystic disease and fibroadenoma of the breast: Natural history and relation to breast cancer risk. Br Med Bull 47(2):258–271.
Donegan, W. L. 2002. Common benign conditions of the breast. In Cancer of the breast, 5th ed. Edited by W. L. Donegan and J. S. Spratt. Philadelphia, PA: Saunders.
Duffy, S. W., E. Lynge, H. Jonsson, S. Ayyaz, and A. H. Olsen. 2008. Complexities in the estimation of overdiagnosis in breast cancer screening. Br J Cancer 99(7):1176–1178.
Easton, D. F., K. A. Pooley, A. M. Dunning, P. D. Pharoah, D. Thompson, D. G. Ballinger, J. P. Struewing, J. Morrison, et al. 2007. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 447(7148):1087–1093.
Eaton, D. L., and E. P. Gallagher. 1994. Mechanisms of aflatoxin carcinogenesis. Annu Rev Pharmacol Toxicol 34:135–172.
Eaton, D. L., and C. D. Klaason. 1996. Principles of toxicology. In Casarett & Doull’s toxicology: The basic science of poisons, 5th ed. Edited by M. J. Wonsiewicz and L. A. Sheinis. New York: McGraw Hill.
Eide, G. E. 2008. Attributable fractions for partitioning risk and evaluating disease prevention: A practical guide. Clin Respir J 2(Suppl 1):92–103.
Eide, G. E., and I. Heuch. 2001. Attributable fractions: Fundamental concepts and their visualization. Stat Methods Med Res 10(3):159–193.
El-Wakeel, H., and H. C. Umpleby. 2003. Systematic review of fibroadenoma as a risk factor for breast cancer. Breast 12(5):302–307.
EPA (Environmental Protection Agency). 1998a. Health effects test guidelines. OPPTS 870.4200: Carcinogenicity. EPA 712–C–98–211. Washington, DC: Government Printing Office. http://hero.epa.gov/index.cfm?action=search.view&reference_ID=6378 (accessed November 16, 2011).
EPA. 1998b. High production volume (HPV) chemicals and SIDS testing. http://www.epa.gov/HPV/pubs/general/hazchem.htm (accessed June 15, 2011).
EPA. 2006. An inventory of sources and environmental releases of dioxin-like compounds in the U.S. for the years 1987, 1995, and 2000 (Final, Nov 2006). http://cfpub.epa.gov/ncea/cfm/recordisplay.cfm?deid=159286 (accessed November 2, 2011).
EPA. 2010. Polychlorinated biphenyls (PCBs). http://www.epa.gov/epawaste/hazard/tsd/pcbs/pubs/about.htm (accessed November 17, 2011).
EPA. 2011. Tox21. http://www.epa.gov/ncct/Tox21/ (accessed November 17, 2011).
Esserman, L., Y. Shieh, and I. Thompson. 2009. Rethinking screening for breast cancer and prostate cancer. JAMA 302(15):1685–1692.
Etique, N., D. Chardard, A. Chesnel, J. L. Merlin, S. Flament, and I. Grillier-Vuissoz. 2004. Ethanol stimulates proliferation, ERα and aromatase expression in MCF-7 human breast cancer cells. Int J Mol Med 13(1):149–155.
FDA (Food and Drug Administration). 1997. Guidance for industry: S1B testing for carcinogenicity of pharmaceuticals. Rockville, MD: FDA. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm074916.pdf (accessed November 17, 2011).
Fenton, S. E. 2006. Endocrine-disrupting compounds and mammary gland development: Early exposure and later life consequences. Endocrinology 147(6 Suppl):S18–S24.
Fenton, S. E., J. T. Hamm, L. S. Birnbaum, and G. L. Youngblood. 2002. Persistent abnormalities in the rat mammary gland following gestational and lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Toxicol Sci 67(1):63–74.
Fernandez, I., P. Touraine, and V. Goffin. 2010. Prolactin and human tumourogenesis. J Neuroendocrinol 22(7):771–777.
Fisher, R. A. 1958a. Cancer and smoking. Nature 182(4635):596.
Fisher, R. A. 1958b. Lung cancer and cigarettes. Nature 182(4628):108.
Foster, P. 2011. NTP’s modified one-generation reproduction study Board of Scientific Counselor’s meeting. http://ntp.niehs.nih.gov/ntp/About_NTP/BSC/2011/April/MOGDesign.pdf (accessed November 13, 2011).
Fritz, W. A., L. Coward, J. Wang, and C. A. Lamartiniere. 1998. Dietary genistein: Perinatal mammary cancer prevention, bioavailability and toxicity testing in the rat. Carcinogenesis 19(12):2151–2158.
Gail, M. H., L. A. Brinton, D. P. Byar, D. K. Corle, S. B. Green, C. Schairer, and J. J. Mulvihill. 1989. Projecting individualized probabilities of developing breast cancer for white females who are being examined annually. J Natl Cancer Inst 81(24):1879–1886.
Gillette, R. W. 1976. Mouse mammary tumor virus as a model for viral carcinogenesis. J Toxicol Environ Health 1(4):545–550.
Goehring, C., and A. Morabia. 1997. Epidemiology of benign breast disease, with special attention to histologic types. Epidemiol Rev 19(2):310–327.
Gohlke, J. M., R. Thomas, Y. Zhang, M. C. Rosenstein, A. P. Davis, C. Murphy, K. G. Becker, C. J. Mattingly, and C. J. Portier. 2009. Genetic and environmental pathways to complex diseases. BMC Syst Biol 3:46.
Goodman, M., M. Kelsh, K. Ebi, J. Iannuzzi, and B. Langholz. 2002. Evaluation of potential confounders in planning a study of occupational magnetic field exposure and female breast cancer. Epidemiology 13(1):50–58.
Gordis, L. 2000. Epidemiology, 2nd ed. Philadelphia, PA: W. B. Saunders Company.
Gøtzsche, P. C., and M. Nielsen. 2006. Screening for breast cancer with mammography. Cochrane Database Syst Rev (4):CD001877.
Gøtzsche, P. C., K. J. Jørgensen, J. Maehlen, and P. H. Zahl. 2009. Estimation of lead time and overdiagnosis in breast cancer screening. Br J Cancer 100(1):219.
Greenland, S., J. Pearl, and J. M. Robins. 1999. Causal diagrams for epidemiologic research. Epidemiology 10(1):37–48.
Guray, M., and A. A. Sahin. 2006. Benign breast diseases: Classification, diagnosis, and management. Oncologist 11(5): 435–449.
Haseman, J. K., J. R. Hailey, and R. W. Morris. 1998. Spontaneous neoplasm incidences in Fischer 344 rats and B6C3F1 mice in two-year carcinogenicity studies: A National Toxicology Program update. Toxicol Pathol 26(3):428–441.
Heinonen, O. P., S. Shapiro, L. Tuominen, and M. I. Turunen. 1974. Reserpine use in relation to breast cancer. Lancet 2(7882):675–677.
Hellmold, H., T. Rylander, M. Magnusson, E. Reihner, M. Warner, and J. A. Gustafsson. 1998. Characterization of cytochrome P450 enzymes in human breast tissue from reduction mammaplasties. J Clin Endocrinol Metab 83(3):886–895.
Henderson, T. O., A. Amsterdam, S. Bhatia, M. M. Hudson, A. T. Meadows, J. P. Neglia, L. R. Diller, L. S. Constine, et al. 2010. Systematic review: Surveillance for breast cancer in women treated with chest radiation for childhood, adolescent, or young adult cancer. Ann Intern Med 152(7):444–455; W144–W154.
Hernαn, M. A., S. Hernandez-Diaz, M. M. Werler, and A. A. Mitchell. 2002. Causal knowledge as a prerequisite for confounding evaluation: An application to birth defects epidemiology. Am J Epidemiol 155(2):176-184.
Herschkowitz, J. I., W. Zhao, M. Zhang, J. Usary, G. Murrow, D. Edwards, J. Knezevic, S. B. Greene, et al. 2011. Comparative oncogenomics identifies breast tumors enriched in functional tumor-initiating cells. Proc Natl Acad Sci U S A. June 1. [Epub ahead of print].
Hilakivi-Clarke, L., E. Cho, I. Onojafe, M. Raygada, and R. Clarke. 1999a. Maternal exposure to genistein during pregnancy increases carcinogen-induced mammary tumorigenesis in female rat offspring. Oncol Rep 6(5):1089–1095.
Hilakivi-Clarke, L., I. Onojafe, M. Raygada, E. Cho, T. Skaar, I. Russo, and R. Clarke. 1999b. Prepubertal exposure to zearalenone or genistein reduces mammary tumorigenesis. Br J Cancer 80(11):1682–1688.
Hilakivi-Clarke, L., A. Cabanes, S. de Assis, M. Wang, G. Khan, W. J. Shoemaker, and R. G. Stevens. 2004. In utero alcohol exposure increases mammary tumorigenesis in rats. Br J Cancer 90(11):2225–2231.
Horwitz, R. I., and A. R. Feinstein. 1985. Exclusion bias and the false relationship of reserpine and breast cancer. Arch Intern Med 145(10):1873–1875.
Howell, A., A. H. Sims, K. R. Ong, M. N. Harvie, D. G. Evans, and R. B. Clarke. 2005. Mechanisms of disease: Prediction and prevention of breast cancer—cellular and molecular interactions. Nat Clin Pract Oncol 2(12):635–646.
Huang, Z., M. J. Fasco, H. L. Figge, K. Keyomarsi, and L. S. Kaminsky. 1996. Expression of cytochromes P450 in human breast tissue and tumors. Drug Metab Dispos 24(8):899–905.
Huff, J. E., S. L. Eustis, and J. K. Haseman. 1989. Occurrence and relevance of chemically induced benign neoplasms in long-term carcinogenicity studies. Cancer Metastasis Rev 8(1):1–22.
Huff, J., J. Cirvello, J. Haseman, and J. Bucher. 1991. Chemicals associated with site-specific neoplasia in 1394 long-term carcinogenesis experiments in laboratory rodents. Environ Health Perspect 93:247–270.
Hunter, D. J., P. Kraft, K. B. Jacobs, D. G. Cox, M. Yeager, S. E. Hankinson, S. Wacholder, Z. Wang, et al. 2007. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet 39(7):870–874.
Hunter, D. J., D. Altshuler, and D. J. Rader. 2008. From Darwin’s finches to canaries in the coal mine—mining the genome for new biology. N Engl J Med 358(26):2760–2763.
IARC (International Agency for Research on Cancer). 1987. Diethylstilbestrol. In Overall evaluations of carcinogenicity: An updating of IARC Monographs volumes 1 to 42. Lyon, France: IARC.
Ioannides, C. 2002. Enzyme systems that metabolise drugs and other xenobiotics. New York: John Wiley and Sons.
Jacobson, E. M., E. R. Hugo, D. C. Borcherding, and N. Ben-Jonathan. 2011. Prolactin in breast and prostate cancer: Molecular and genetic perspectives. Discov Med 11(59):315–324.
Jenkins, S., N. Raghuraman, I. Eltoum, M. Carpenter, J. Russo, and C. A. Lamartiniere. 2009. Oral exposure to bisphenol A increases dimethylbenzanthracene-induced mammary cancer in rats. Environ Health Perspect 117(6):910–915.
Jo, W. K., C. P. Weisel, and P. J. Lioy. 1990. Chloroform exposure and the health risk associated with multiple uses of chlorinated tap water. Risk Anal 10(4):581–585.
Kabat, G. C., J. G. Jones, N. Olson, A. Negassa, C. Duggan, M. Ginsberg, R. A. Kandel, A. G. Glass, and T. E. Rohan. 2010. A multi-center prospective cohort study of benign breast disease and risk of subsequent breast cancer. Cancer Causes Control 21(6):821–828.
Kacew, S., and M. F. Festing. 1996. Role of rat strain in the differential sensitivity to pharmaceutical agents and naturally occurring substances. J Toxicol Environ Health 47(1):1–30.
Kacew, S., Z. Ruben, and R. F. McConnell. 1995. Strain as a determinant factor in the differential responsiveness of rats to chemicals. Toxicol Pathol 23(6):701–714; discussion 714–715.
Kanhai, R. C., J. J. Hage, E. Bloemena, P. J. van Diest, and R. B. Karim. 1999. Mammary fibroadenoma in a male-to-female transsexual. Histopathology 35(2):183–185.
Khan, G., P. Penttinen, A. Cabanes, A. Foxworth, A. Chezek, K. Mastropole, B. Yu, A. Smeds, et al. 2007. Maternal flaxseed diet during pregnancy or lactation increases female rat offspring’s susceptibility to carcinogen-induced mammary tumorigenesis. Reprod Toxicol 23(3):397–406.
Klepeis, N. E., A. M. Tsang, and J. V. Behar. 1995. Analysis of the national human activity pattern survey (NHAPS) respondents from a standpoint of exposure assessment. National Exposure Research Laboratory Office of Research and Development. Las Vegas, NV: EPA. http://www.exposurescience.org/pub/reports/NHAPS_Report1.pdf (accessed November 3, 2011).
Kochukov, M. Y., Y. J. Jeng, and C. S. Watson. 2009. Alkylphenol xenoestrogens with varying carbon chain lengths differentially and potently activate signaling and functional responses in GH3/B6/F10 somatomammotropes. Environ Health Perspect 117(5):723–730.
Komenaka, I. K., K. D. Miller, and G. W. Sledge. 2006. Male breast cancer. In Textbook of uncommon cancer, 3rd ed. Edited by D. Raghavan, M. L. Brecher, D. H. Johnson, N. J. Meropol, P. L. Moots, P. G. Rose, and I. A. Mayer. Hoboken, NJ: Wiley.
Kramer, B. S., and J. M. Croswell. 2009. Cancer screening: The clash of science and intuition. Annu Rev Med 60:125–137.
Kramer, B. S., and J. Croswell. 2010. Limitations and pitfalls in the early detection of cancer: History, current concerns, and the future. AUA Update Series 29(Lesson 7):65–76.
Krewski, D., M. E. Andersen, E. Mantus, and L. Zeise. 2009. Toxicity testing in the 21st century: Implications for human health risk assessment. Risk Anal 29(4):474–479.
Kuiper, A. 2005. Pathogenesis and progression of fibroepithelial breast tumors. Ph.D. diss., University of Utrecht, Netherlands.
La Merrill, M., R. Harper, L. S. Birnbaum, R. D. Cardiff, and D. W. Threadgill. 2010. Maternal dioxin exposure combined with a diet high in fat increases mammary cancer incidence in mice. Environ Health Perspect 118(5):596–601.
Laden, F., G. Collman, K. Iwamoto, A. J. Alberg, G. S. Berkowitz, J. L. Freudenheim, S. E. Hankinson, K. J. Helzlsouer, et al. 2001. 1,1-dichloro-2,2-bis(p-chlorophenyl)ethylene and polychlorinated biphenyls and breast cancer: Combined analysis of five U.S. studies. J Natl Cancer Inst 93(10):768–776.
Laden, F., N. Ishibe, S. E. Hankinson, M. S. Wolff, D. M. Gertig, D. J. Hunter, and K. T. Kelsey. 2002. Polychlorinated biphenyls, cytochrome P450 1A1, and breast cancer risk in the Nurses’ Health Study. Cancer Epidemiol Biomarkers Prev 11(12):1560–1565.
Lanari, C., C. A. Lamb, V. T. Fabris, L. A. Helguero, R. Soldati, M. C. Bottino, S. Giulianelli, J. P. Cerliani, et al. 2009. The MPA mouse breast cancer model: Evidence for a role of progesterone receptors in breast cancer. Endocr Relat Cancer 16(2):333–350.
Land, C. E., M. Tokunaga, K. Koyama, M. Soda, D. L. Preston, I. Nishimori, and S. Tokuoka. 2003. Incidence of female breast cancer among atomic bomb survivors, Hiroshima and Nagasaki, 1950–1990. Radiat Res 160(6):707–717.
Langholz, B. 2001. Factors that explain the power line configuration wiring code–childhood leukemia association: What would they look like? Bioelectromagnetics (Suppl 5):S19–S31.
Langholz, B., D. C. Thomas, M. Stovall, S. A. Smith, J. D. Boice, Jr., R. E. Shore, L. Bernstein, C. F. Lynch, et al. 2009. Statistical methods for analysis of radiation effects with tumor and dose location-specific information with application to the WECARE study of asynchronous contralateral breast cancer. Biometrics 65(2):599–608.
Li, D., W. Zhang, A. A. Sahin, and W. N. Hittelman. 1999. DNA adducts in normal tissue adjacent to breast cancer: A review. Cancer Detect Prev 23(6):454–462.
Li, Y., R. C. Millikan, D. A. Bell, L. Cui, C. K. Tse, B. Newman, and K. Conway. 2005. Polychlorinated biphenyls, cytochrome P450 1A1 (CYP1A1) polymorphisms, and breast cancer risk among African American women and white women in North Carolina: A population-based case–control study. Breast Cancer Res 7(1):R12–R18.
Lilienfeld, A. M., and D. E. Lilienfeld. 1980. Laying the foundations: The epidemiologic approach to disease. In Foundations of Epidemiology. New York: Oxford University Press. Pp. 3–22.
Madigan, M. P., R. G. Ziegler, J. Benichou, C. Byrne, and R. N. Hoover. 1995. Proportion of breast cancer cases in the United States explained by well-established risk factors. J Natl Cancer Inst 87(22):1681–1685.
Makris, S. L. 2011. Current assessment of the effects of environmental chemicals on the mammary gland in guideline rodent studies by the U.S. Environmental Protection Agency (U.S. EPA), Organisation For Economic Co-Operation And Development (OECD), and National Toxicology Program (NTP). Environ Health Perspect 119(8):1047–1052.
Manolio, T. A. 2010. Genomewide association studies and assessment of the risk of disease. N Engl J Med 363(2):166–176.
Manolio, T. A., F. S. Collins, N. J. Cox, D. B. Goldstein, L. A. Hindorff, D. J. Hunter, M. I. McCarthy, E. M. Ramos, et al. 2009. Finding the missing heritability of complex diseases. Nature 461(7265):747–753.
Markopoulos, C., E. Kouskos, D. Mantas, K. Kontzoglou, K. Antonopoulou, Z. Revenas, and V. Kyriakou. 2004. Fibroadenomas of the breast: Is there any association with breast cancer? Eur J Gynaecol Oncol 25(4):495–497.
Masson, L. F., L. Sharp, S. C. Cotton, and J. Little. 2005. Cytochrome P-450 1A1 gene polymorphisms and risk of breast cancer: A HuGE review. Am J Epidemiol 161(10):901–915.
Medina, D. 2007. Chemical carcinogenesis of rat and mouse mammary glands. Breast Dis 28:63–68.
Medina, D. 2010. Of mice and women: A short history of mouse mammary cancer research with an emphasis on the pardigms inspired by the transplantation method. Cold Spring Harb Perspect Biol 2(10):a004523.
Meranze, D. R., M. Gruenstein, and M. B. Shimkin. 1969. Effect of age and sex on the development of neoplasms in Wistar rats receiving a single intragastric instillation of 7,13-dimethylbenz(a)anthracene. Int J Cancer 4(4):480–486.
Morrison, A. 1985. Screening in chronic disease. New York: Oxford University Press.
Morrison, J. H., R. D. Brinton, P. J. Schmidt, and A. C. Gore. 2006. Estrogen, menopause, and the aging brain: How basic neuroscience can inform hormone therapy in women. J Neurosci 26(41):10332–10348.
Moysich, K. B., P. G. Shields, J. L. Freudenheim, E. F. Schisterman, J. E. Vena, P. Kostyniak, H. Greizerstein, J. R. Marshall, et al. 1999. Polychlorinated biphenyls, cytochrome P4501A1 polymorphism, and postmenopausal breast cancer risk. Cancer Epidemiol Biomarkers Prev 8(1):41–44.
MRFIT Research Group. 1982. Multiple risk factor intervention trial. Risk factor changes and mortality results. Multiple Risk Factor Intervention Trial Research Group. JAMA 248(12):1465–1477.
Narod, S. A., and K. Offit. 2005. Prevention and management of hereditary breast cancer. J Clin Oncol 23(8):1656–1663.
NCI (National Cancer Institute). 2010. SEER cancer statistics review, 1975–2007. Edited by S. F. Altekruse, C. L. Kosary, M. Krapcho, N. Neyman, R. Aminou, W. Waldron, J. Ruhl, N. Howlader, Z. Tatalovich, H. Cho, A. Mariotto, M. P. Eisner, D. R. Lewis, K. Cronin, H. S. Chen, E. J. Feuer, D. G. Stinchcomb, and B. K. Edwards. Bethesda, MD: NCI. http://seer.cancer.gov/csr/1975_2007/ (accessed January 6, 2011).
Nebert, D. W., and T. P. Dalton. 2006. The role of cytochrome P450 enzymes in endogenous signalling pathways and environmental carcinogenesis. Nat Rev Cancer 6(12):947–960.
Nelson, D. R., D. C. Zeldin, S. M. Hoffman, L. J. Maltais, H. M. Wain, and D. W. Nebert. 2004. Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics 14(1):1–18.
Nguyen, D. H., H. A. Oketch-Rabah, I. Illa-Bochaca, F. C. Geyer, J. S. Reis-Filho, J. H. Mao, S. A. Ravani, J. Zavadil, et al. 2011. Radiation acts on the microenvironment to affect breast carcinogenesis by distinct mechanisms that decrease cancer latency and affect tumor type. Cancer Cell 19(5):640–651.
Noolkar, A., D. G. Mote, and S. D. Deshpande. 2010. Case report: A case of primary bilateral fibroadenoma of breasts in a male 20 years of age. In Proceedings of the World Medical Conference, Malta, September 15–17, 2010. Edited by V. V. Sumbayev, A. Pica, S.-P. Luh, H. K. Lim, K. Natori, and S. Meshitsuka. WSEAS Press. http://www.wseas.us/e-library/conferences/2010/Malta/MEDICAL/MEDICAL-29.pdf (accessed November 11, 2011).
NRC (National Research Council). 2007. Toxicity testing in the 21st century: A vision and a strategy. Washington, DC: The National Academies Press.
NTP (National Toxicology Program). 2008. NTP historical controls report, all routes and vehicles: Harlan Sprague-Dawley rats. http://ntp.niehs.nih.gov/ntp/Historical_Controls/NTP2000_2009/2008_HC_Report_SD_Rats_ByRoute.pdf (accessed December 14, 2011).
NTP. 2010a. NTP historical controls report, all routes and vehicles: Rats. http://ntp.niehs.nih.gov/ntp/Historical_Controls/NTP2000_2010/2010-03-22-HIST-RatsAllRoutes.pdf (accessed December 22, 2011).
NTP. 2010b. NTP historical controls report, all routes and vehicles: Mice. http://ntp.niehs.nih.gov/ntp/Historical_Controls/NTP2000_2010/2010-03-22-HIST-MiceAllRoutes.pdf (accessed December 22, 2011).
NTP. 2010c. Toxicology/carcinogenicity. http://ntp.niehs.nih.gov/?objectid=72015DAF-BDB7-CEBA-F9A7F9CAA57DD7F5 (accessed November 13, 2011).
Park, J. H., S. Wacholder, M. H. Gail, U. Peters, K. B. Jacobs, S. J. Chanock, and N. Chatterjee. 2010. Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat Genet 42(7):570–575.
Patel, C. J., J. Bhattacharya, and A. J. Butte. 2010. An environment-wide association study (EWAS) on type 2 diabetes mellitus. PLoS One 5(5):e10746.
Paul, A. G., K. C. Jones, and A. J. Sweetman. 2009. A first global production, emission, and environmental inventory for perfluorooctane sulfonate. Environ Sci Technol 43(2): 386–392.
Pereira, P. C., A. F. Vicente, A. S. Cabrita, and M. F. Mesquita. 2009. The influence of olive oil on Sprague Dawley rats DMBA-induced mammary tumors. International Journal of Cancer Research 5(4):144–152.
Perera, M. I., H. W. Kunz, T. J. Gill, 3rd, and H. Shinozuka. 1986. Enhancement of induction of intestinal adenocarcinomas by cyclosporine in rats given a single dose of N-methyl N-nitrosourea. Transplantation, 42:297–302.
Perou, C. M., T. Sorlie, M. B. Eisen, M. van de Rijn, S. S. Jeffrey, C. A. Rees, J. R. Pollack, D. T. Ross, et al. 2000. Molecular portraits of human breast tumours. Nature 406(6797): 747–752.
Petersen, M. L., S. E. Sinisi, and M. J. van der Laan. 2006. Estimation of direct causal effects. Epidemiology 17(3):276–284.
Preston, D. L., E. Ron, S. Tokuoka, S. Funamoto, N. Nishi, M. Soda, K. Mabuchi, and K. Kodama. 2007. Solid cancer incidence in atomic bomb survivors: 1958–1998. Radiat Res 168(1):1–64.
Rappaport, S. M. 2011. Implications of the exposome for exposure science. J Expo Sci Environ Epidemiol 21(1):5–9.
Reeves, G. K., R. C. Travis, J. Green, D. Bull, S. Tipper, K. Baker, V. Beral, R. Peto, et al. 2010. Incidence of breast cancer and its subtypes in relation to individual and multiple low-penetrance genetic susceptibility loci. JAMA 304(4):426–434.
Ren, X., X. Zhang, A. S. Kim, A. M. Mikheev, M. Fang, R. C. Sullivan, R. E. Bumgarner, and H. Zarbl. 2008. Comparative genomics of susceptibility to mammary carcinogenesis among inbred rat strains: Role of reduced prolactin signaling in resistance of the Copenhagen strain. Carcinogenesis 29(1):177–185.
Rockhill, B., B. Newman, and C. Weinberg. 1998. Use and misuse of population attributable fractions. Am J Public Health 88(1):15–19.
Rose, G. 1985. Sick individuals and sick populations. Int J Epidemiol 14(1):32–38.
Rosen, P. R. 2001. Benign proliferative lesions of the male breast. In Rosen’s breast pathology, 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins.
Rosen, P. R. 2009. Fibroepithelial neoplasms. In Rosen’s breast pathology, 3rd ed. Philadelphia, PA: Wolters Kluwer Health/Lippincott Williams & Wilkins.
Rothman, K. J., S. Greenland, and T. L. Lash. 2008. Modern epidemiology, 3rd ed. Philadelphia, PA: Lippincott Williams & Wilkins.
Rudel, R. A., K. R. Attfield, J. N. Schifano, and J. G. Brody. 2007. Chemicals causing mammary gland tumors in animals signal new directions for epidemiology, chemicals testing, and risk assessment for breast cancer prevention. Cancer 109(12 Suppl):2635–2666.
Rudel, R. A., R. E. Dodson, L. J. Perovich, R. Morello-Frosch, D. E. Camann, M. M. Zuniga, A. Y. Yau, A. C. Just, et al. 2010. Semivolatile endocrine-disrupting compounds in paired indoor and outdoor air in two northern California communities. Environ Sci Technol 44(17):6583–6590.
Rudel, R. A., S. E. Fenton, J. M. Ackerman, S. Y. Euling, and S. L. Makris. 2011. Environmental exposures and mammary gland development: State of the science, public health implications, and research recommendations. Environ Health Perspect 119(8):1053–1061.
Russo, I. H., and J. Russo. 1996. Mammary gland neoplasia in long-term rodent studies. Environ Health Perspect 104(9):938–967.
Sax, S. N., D. H. Bennett, S. N. Chillrud, J. Ross, P. L. Kinney, and J. D. Spengler. 2006. A cancer risk assessment of inner-city teenagers living in New York City and Los Angeles. Environ Health Perspect 114(10):1558–1566.
Seidman, H., S. D. Stellman, and M. H. Mushinski. 1982. A different perspective on breast cancer risk factors: Some implications of the nonattributable risk. CA Cancer J Clin 32(5):301–313.
Sergeev, P. V., T. V. Ukhina, K. G. Gurevich, and N. L. Shimanovskii. 2001. On the mechanisms of various dose–effect relationships for estrogens. Pharm Chem J 35(9):468–470.
Shin, S. J., and P. P. Rosen. 2007. Bilateral presentation of fibroadenoma with digital fibroma-like inclusions in the male breast. Arch Pathol Lab Med 131(7):1126–1129.
Stacey, S. N., A. Manolescu, P. Sulem, T. Rafnar, J. Gudmundsson, S. A. Gudjonsson, G. Masson, M. Jakobsdottir, et al. 2007. Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet 39(7): 865–869.
Steenland, K., and B. Armstrong. 2006. An overview of methods for calculating the burden of disease due to specific risk factors. Epidemiology 17(5):512–519.
Stovall, M., R. Weathers, C. Kasper, S. A. Smith, L. Travis, E. Ron, and R. Kleinerman. 2006. Dose reconstruction for therapeutic and diagnostic radiation exposures: Use in epidemiological studies. Radiat Res 166(1 Pt 2):141–157.
Stovall, M., S. A. Smith, B. M. Langholz, J. D. Boice, Jr., R. E. Shore, M. Andersson, T. A. Buchholz, M. Capanu, et al. 2008. Dose to the contralateral breast from radiotherapy and risk of second primary breast cancer in the WECARE study. Int J Radiat Oncol Biol Phys 72(4):1021–1030.
Sudaryanto, A., N. Kajiwara, S. Takahashi, S. Muawanah, and S. Tanabe. 2008. Geographical distribution and accumulation features of PBDEs in human breast milk from Indonesia. Environ Pollut 151(1):130–138.
Thayer, K. A., and P. M. Foster. 2007. Workgroup report: National Toxicology Program workshop on hormonally induced reproductive tumors—relevance of rodent bioassays. Environ Health Perspect 115(9):1351–1356.
Travis, R. C., G. K. Reeves, J. Green, D. Bull, S. J. Tipper, K. Baker, V. Beral, R. Peto, et al. 2010. Gene–environment interactions in 7610 women with breast cancer: Prospective evidence from the Million Women Study. Lancet 375(9732):2143–2151.
Turnbull, C., S. Ahmed, J. Morrison, D. Pernet, A. Renwick, M. Maranian, S. Seal, M. Ghoussaini, et al. 2010. Genome-wide association study identifies five new breast cancer susceptibility loci. Nat Genet 42(6):504–507.
Turpin, B. J., C. P. Weisel, M. Morandi, S. Colome, T. Stock, S. Eisenreich, and B. Buckley. 2007. Relationships of indoor, outdoor, and personal air (RIOPA): Part II. Analyses of concentrations of particulate matter species. HEI Research Report 130; NUATRC Research Report 10. Boston MA: Health Effects Institute, and Houston TX: Mickey Leland National Urban Air Toxics Research Center. http://pubs.healtheffects.org/getfile.php?u=379 (accessed November 8, 2011).
Ullrich, R. L., N. D. Bowles, L. C. Satterfield, and C. M. Davis. 1996. Strain-dependent susceptibility to radiation-induced mammary cancer is a result of differences in epithelial cell sensitivity to transformation. Radiat Res 146(3):353–355.
Uter, W., and A. Pfahlberg. 2001. The application of methods to quantify attributable risk in medical practice. Stat Methods Med Res 10(3):231–237.
Varghese, J. S., and D. F. Easton. 2010. Genome-wide association studies in common cancers—what have we learnt? Curr Opin Genet Dev 20(3):201–209.
Wacholder, S., P. Hartge, R. Prentice, M. Garcia-Closas, H. S. Feigelson, W. R. Diver, M. J. Thun, D. G. Cox, et al. 2010a. Performance of common genetic variants in breast-cancer risk models. N Engl J Med 362(11):986–993.
Wacholder, S., P. Hartge, R. Prentice, M. Garcia-Closas, H. S. Feigelson, W. R. Diver, M. J. Thun, D. G. Cox, et al. 2010b. Supplement to: Performance of common genetic variants in breast-cancer risk models. N Engl J Med 362(11):986–993.
Ward, T. J., H. Underberg, D. Jones, R. F. Hamilton, Jr., and E. Adams. 2009. Indoor/ambient residential air toxics results in rural western Montana. Environ Monit Assess 153(1–4):119–126.
Watson, C. S., N. N. Bulayeva, A. L. Wozniak, and R. A. Alyea. 2007. Xenoestrogens are potent activators of nongenomic estrogenic responses. Steroids 72(2):124–134.
Welch, H. G., and W. C. Black. 2010. Overdiagnosis in cancer. J Natl Cancer Inst 102(9): 605–613.
Welch, H. G., L. M. Schwartz, and S. Woloshin. 2000. Are increasing 5-year survival rates evidence of success against cancer? JAMA 283(22):2975–2978.
Welshons, W. V., K. A. Thayer, B. M. Judy, J. A. Taylor, E. M. Curran, and F. S. vom Saal. 2003. Large effects from small exposures. Mechanisms for endocrine-disrupting chemicals with estrogenic activity. Environ Health Perspect 111(8):994–1006.
Yin, W., and A. C. Gore. 2006. Neuroendocrine control of reproductive aging: Roles of GnRH neurons. Reproduction 131(3):403–414.
Zackrisson, S., I. Andersson, L. Janzon, J. Manjer, and J. P. Garne. 2006. Rate of over-diagnosis of breast cancer 15 years after end of Malmo mammographic screening trial: Follow-up study. BMJ 332(7543):689–692.
Zheng, W., J. Long, Y. T. Gao, C. Li, Y. Zheng, Y. B. Xiang, W. Wen, S. Levy, et al. 2009. Genome-wide association study identifies a new breast cancer susceptibility locus at 6q25.1. Nat Genet 41(3):324–328.
This page intentionally left blank.






























































