
A Shared Responsibility for Improving Health IT Safety
As discussed in Chapter 2, the use of health IT in some areas has significantly improved the quality of health care and reduced medical errors. Continuing to use paper medical records can place patients at unnecessary risk for harm and substantially constrain the country’s ability to reform the health care system. However, there are clearly cases in which harm has occurred associated with new health IT. The committee believes safer health care is possible in complex, dynamic environments—which are the rule in health care—only when achieving and maintaining safety is given a high priority.
Achieving the desired reduction in harm will depend on a number of factors, including how the technology is designed, how it is implemented, how well it fits into clinical workflow, how it supports informed decision making by both patients and providers, and whether it is safe and reliable. An environment of safer health IT can be created if both the public and private sectors acknowledge that safety is a shared responsibility. Actions are needed to correct the market and commit to ensuring the safety of health IT. A better understanding and acknowledgement of the risks associated with health IT and its use, as well as how to maximize the benefits, are needed. An example of a new kind of error that can occur with IT which did not occur previously is the “adjacency error,” in which a provider selects an item next to the one intended from a pulldown menu, for example picking “penicillamine” instead of “penicillin.” Such errors occur in many products, but effective solutions have not yet generally been fielded. This chapter details the actions to be taken by both the public and private sectors that the committee believes will be necessary for the creation of an environment
in which IT improves safety overall, and the new problems created by health IT are minimized.
THE ROLE OF THE PRIVATE SECTOR: PROMOTING SHARED LEARNING ENVIRONMENTS
This chapter broadly defines the private sector to include health IT vendors, insurers, and the organizations that support each of these groups (e.g., professional societies). Health care organizations, health professionals, and patients and their families are also considered part of the private sector. The public sector generally refers to the government. Operationally, the line between the private and public sectors is not completely clear, because some organizations operate in both sectors.
The current environment in which health IT is designed and used does not adequately protect patient safety. However, the private sector has the ability to drive innovation and creativity, generating the tools to deliver the best possible health care and directly improve safety. In this regard, the private sector has the most direct responsibility to realign the market, but it will need support from the public sector.
The complexity and dynamism of health IT requires that private-sector entities work together through shared learning to improve patient safety. Manufacturers and health professionals have to communicate their capabilities and needs to each other to facilitate the design of health IT in ways that achieve maximum usability and safety. Patients and their families need to be able to interact seamlessly with health professionals through patient engagement tools. Health care organizations ought to share lessons learned with each other to avoid common patient safety risks as they adopt highly complex health IT products.
However, today’s reality is that the private sector currently consists of a broad variety of stakeholders lacking a uniform approach, and potentially misaligned goals. The track record of the private sector in responding to new safety issues created by IT is mixed. Although nearly all stakeholders would endorse the broad goals of improving the quality and safety of patient care, many stakeholders (particularly vendors) are faced with competing priorities, including maximizing profits and maintaining a competitive edge, which can limit shared learning, and have adverse consequences for patient safety. Shared learning about safety risks and mitigation strategies for safer health IT among users, vendors, researchers, and other stakeholders, can optimize patient safety and minimize harm.
As discussed in Chapters 4 and 5, there are many opportunities for the private sector to improve safety as it relates to health IT, but to date, little action has been taken. Insufficient action by the private sector to improve patient safety can endanger lives. The private sector must play a major role
in addressing this urgent need to better understand risks and benefits associated with health IT, as well as strategies for improvement and remediation. As it stands now, there is a lack of accountability on the part of vendors, who are generally perceived to shift responsibility and accountability to users through specific contract language (Goodman et al., 2010). As a result, the committee believes a number of critical gaps in knowledge need to be addressed immediately, including the lack of comprehensive mechanisms for identifying patient safety risks, measuring health IT safety, ensuring safe implementation, and educating and training users.
Developing a System for Identifying Patient Safety Risks
The committee believes that transparency, characterized by developing, identifying, and sharing evidence on risks to patient safety, is essential to a properly functioning market where users would have the ability to choose a product that best suits their needs and the needs of their patients. However, the committee found sparse evidence pertaining to the volume and types of patient safety risks related to health IT. Indeed, the number of errors reported both anecdotally and in the published literature was lower than the committee anticipated. This led primarily to the sense that potentially harmful situations and adverse events caused by IT were often not recognized and, even when they were recognized, usually not reported. This lack of reported instances of harm is consistent with other areas of patient safety, including paper-based patient records and other manually based care systems, where there is ample evidence that most adverse events are never reported, even when there are robust programs encouraging health professionals to do so (Classen et al., 2011; Cullen et al., 1995).
Information technology can assist organizations in identifying, troubleshooting, and handling health IT-related adverse events. Digital forensic tools (e.g., centralized logging, regular system backups) can be used to record data during system use. After an adverse event occurs, recorded data—such as log-in information, keystrokes, and how information is transported throughout the network—can be used to identify, reconstruct, and understand in detail how an adverse event occurred (NIST, 2006).
Because of the diversity of health IT products and their differing effects on various clinical environments, it is essential that users share detailed information with other users, researchers, and the vendor once information regarding adverse events is identified. Examples of such information include screenshots or descriptions of potentially unsafe processes that could help illustrate how a health IT product threatened patient safety. However, as discussed in Chapter 2, users may fear that sharing this information may violate nondisclosure clauses and vendors’ intellectual property, exposing them to liability and litigation. Although there is little evidence on the
impact of such clauses, the committee believes users may be less likely to share information necessary to improve patient safety, given these clauses.
If it is clearly understood that transparently sharing health IT issues with the public is for the purpose of patient safety, vendors ought to agree to remove such restrictions from contracts and work with users to explicitly define what can be shared, who it can be shared with, and for what purposes. However, to maintain a competitive advantage, many vendors may not be motivated to allow users to disclose patient safety-related risks associated with their health IT products. Many vendors place these clauses within the boilerplate language, and in the absence of comprehensive legal review, users may not even realize these restrictions exist when signing their contracts.1 If more users carefully search for such clauses and negotiate terms that allow them to share information related to patient safety risks, vendors may be more likely to exclude such clauses. Furthermore, if it were easier to know which vendors had standard contracts that allowed for sharing, users might be more likely to select those vendors. However, users—particularly smaller organizations—are not part of a “cohesive community” with the legal expertise or knowledge to negotiate such changes. Therefore, the committee believes the Secretary of the Department of Health and Human Services (HHS) should provide tools to motivate vendors and empower users to negotiate contracts that allow for sharing of patient safety-related details and improved transparency. The Secretary ought to investigate what other tools and authorities would be required to ensure the free exchange of patient safety-related information.
Recommendation 2: The Secretary of HHS should ensure insofar as possible that health IT vendors support the free exchange of information about health IT experiences and issues and not prohibit sharing of such information, including details (e.g., screenshots) relating to patient safety.
The committee recognizes that, short of Congressional and regulatory action, the Secretary cannot guarantee how contracts are developed between two private parties. However, the committee views prohibition of the free exchange of information to be the most critical barrier to patient safety and transparency. The committee urges the Secretary to take vigorous steps to restrict contractual language that impedes public sharing of patient safety-related details. Contracts should be developed to allow explicitly for sharing of health IT issues related to patient safety. One method the Secretary could use is to ask the Office of the National Coordinator for Health Information Technology (ONC) to create a list of vendors that satisfy this
![]()
1 Personal communication, E. Belmont, MaineHealth, September 21, 2011.
requirement and/or those that do not. If such a list were available, users could more easily choose vendors that allow patient safety-related details of health IT products to be shared. Having such a list could also motivate vendors to include contractual terms that allow for sharing of patient safety-related details and, as a result, be more competitive to users. The ONC could also consider creating minimum criteria for determining when a contract adequately allows for sharing of patient safety-related details. These criteria need to define the following:
• What situations allow for sharing patient safety-related details of health IT products;
• What content of health IT should be shareable;
• Which stakeholders the information can be shared with, including other users, consumer groups, researchers, and the government; and
• What the limitations of liability are when such information is shared.
Private certification bodies such as the ONC-authorized testing and certification bodies (ONC-ATCBs)2 could also promote the free exchange of patient safety-related information. This could be implemented for example through the creation of a new type of certification that requires this information to be shared.
The Secretary could ask the ONC to develop model contract language that would affirmatively establish the ability of users to provide content and contextual information when reporting an adverse event or unsafe condition. Additionally, HHS could educate users about these contracts and develop guidance for users about what to look for before signing a contract. This education could potentially be done through the ONC’s regional extension centers or the Centers for Medicare & Medicaid Services’s (CMS’s) quality improvement organizations. This effort could also be supported by various professional societies.
To identify how pervasive these clauses are, the Secretary may need to conduct a review of existing contracts. Although the Secretary may not be privy to vendor-purchaser contracts, HHS could conduct a survey or ask vendors to voluntarily share examples of their contract language. Understanding the magnitude of these clauses would be a critical first step.
Once this information is available, comparative user experiences can be made public. There is currently no effective way for users to communicate their experiences with a health IT product. In many other industries, user
![]()
2 ONC-ATCBs as of April 2011 include the Certification Commission for Health Information Technology; Drummond Group, Inc.; ICSALabs; InfoGard Laboratories, Inc.; SLI Globan Solutions; and Surescripts LLC.
reviews appear on online forums and other similar guides, while independent tests are conducted by Consumer Reports and others. These reviews allow users to better understand the products they might be purchasing. Perhaps the more powerful aspect of users being able to rate and compare their experiences with products is the ability to share and report lessons learned. Comparative user experiences for health IT safety need to be created to enhance communication of safety concerns and ways to mitigate potential risks.
To gather objective information about health IT products, researchers should have access to both test versions of software provided by vendors and software already integrated in user organizations. Documentation for health IT products such as user manuals also could be made available to researchers. Resources should be available to share user experiences and other measures of safety specifying data from health IT products. The private sector needs to be a catalyzing force in this area, but governance from the public sector may be required for such tools to be developed.
Recommendation 3: The ONC should work with the private and public sectors to make comparative user experiences across vendors publicly available.
Another way to increase transparency in the private sector is to require reporting of health IT-related adverse events through health care provider accrediting organizations such as The Joint Commission or the National Committee for Quality Assurance (NCQA). Professional associations of providers could also play this role. One of the tools the ONC could provide to facilitate the implementation of Recommendation 3 is the development of a uniform format for making these reports, which could be coordinated through the Common Formats.3 However, it is important to note that a public-sector entity could also lead change in this regard.
Finally, a more robust and comprehensive infrastructure is needed for providing technical assistance to users who may need advice or training to safely implement health IT products. Shared learning between users and vendors in the form of feedback about how well health IT products are working can help improve the focus on safety and usability in the design of health IT products and identification of performance requirements. Tools to foster this feedback in an organized way are needed to promote safety and quality. The learning curve for safely using health IT-assisted care varies widely and technical assistance needs to be provided to users at all levels.
![]()
3 The Common Formats are coordinated by the Agency for Healthcare Research and Quality (AHRQ) in an effort to facilitate standardized reporting of adverse events by creating general definitions and reporting formats for widespread use (AHRQ, 2011a).
Measuring Health IT Safety
Another area the committee identified as necessary for making health IT safer is the development of measures. As is often said in quality improvement, you can only improve what you measure. Currently, few measures address patient safety as it relates to health IT; without these measures, it will be very difficult to develop and test strategies to ensure safe patient care. Although there has been progress in developing general measures of patient safety, the committee concluded that safety measures focusing on the impact of health IT must go beyond traditional safety measures (as discussed in Chapter 3) and are urgently needed.
Measures of health care safety and quality are generally developed by groups such as professional societies and academic researchers and undergo a voluntary consensus process before being adopted for widespread use. During the measure development process, decisions need to be made such as identifying what metrics can be used as an indicator of health IT safety, specifications of the metrics, and the criteria against which measures can be evaluated. Policies for measure ownership and processes for evaluating and maintaining measures will also need to be created. One example of the type of data that are likely to be important would be override rates for important types of safety warnings on alerts and warnings built into electronic health records (EHRs).
The committee believes a consensus-based collaborative effort that would oversee development, application, and evaluation of criteria for measures and best practices of safety of health IT—a Health IT Safety Council—is of vital need. For example, the council could be responsible for identifying key performance aspects of health IT and creating a prioritized agenda for measure development. Given that the process for developing health IT safety metrics would be similar to developing measures of health care safety and quality, a voluntary consensus standards organization would effectively be able to house the recommended council. Because of the ubiquity and complexity of health IT, all health IT stakeholders ought to be involved in the development of such criteria. HHS ought to consider providing the initial funding for the council because the need for measures of safety of health IT is central to all stakeholders. The more costly process of maintaining measures ought to be funded by private-sector entities.
Recommendation 4: The Secretary of HHS should fund a new Health IT Safety Council to evaluate criteria for assessing and monitoring the safe use of health IT and the use of health IT to enhance safety. This council should operate within an existing voluntary consensus standards organization.
One existing organization with a strong history of convening groups and experience with endorsing health care quality and performance measures that could guide this process is the National Quality Forum (NQF). The NQF is a nonprofit organization whose mission is to develop consensus on national priorities and endorse standards for measuring and publicly reporting on health care performance based on measures submitted from measure developers. Of particular note, the NQF hosts eight member councils, whose purposes are to build consensus among council members toward advancing quality measurement and reporting (NQF, 2011). These councils provide a voice to stakeholder groups—including consumers, health professionals, health care organizations, professional organizations (e.g., the American Medical Informatics Association [AMIA]), vendors (e.g., individually and through societies such as the Healthcare Information and Management Systems Society [HIMSS]), and insurers—to identify what types of metrics are needed and the criteria for doing so. Measures should be NQF-endorsed, a process that applies nationally accepted standards and criteria. The NQF could provide guidance in identifying criteria against which to develop health IT safety measures to help gain consensus on the right set of policies.
The ensuing task of developing measures of health IT safety needs to be undertaken by a variety of entities. To accomplish this, some research will be needed for measure development because good measures currently do not exist; these efforts should be supported by the Agency for Healthcare Research and Quality (AHRQ), the National Library of Medicine, and the ONC, as discussed in Recommendation 1. Health care organizations and even vendors could partner with more traditional measurement development organizations, for example the NCQA can create measures that would by default be subjected to the NQF consensus approval process.
Ensuring Safer Implementation
Efforts to safely implement health IT must address three phases: pre- implementation, implementation, and postimplementation of health IT.
Preimplementation
Vendors, with input from users, play the most significant role in the preimplementation phase of health IT. Vendors ought to be able to assert that their products are designed and developed in a way that promotes patient safety. Currently, health IT products are held to few standards with respect to both design and development. Although it is typically the role of standards development organizations such as the American National Standards Institute, the Association for the Advancement of Medical Instrumentation, Health Level 7 (HL7), and the Institute of Electrical and Electronics
Engineers to develop such standards, criteria, and tests, a broader group of stakeholders including patients, users, and vendors should participate in creating safety standards and criteria against which health IT ought to be tested.
Vendors are currently being required by the ONC to meet a specific set of criteria in order for their products to be certified as eligible for use in HHS’s meaningful use program. These criteria relate to clinical functionality, security, and interoperability and may be helpful for but not sufficient to ensure health IT-related safety. The American National Standards Institute, as the body that will accredit organizations that certify health IT, and certification bodies such as the Certification Commission for Healthcare Information Technology serve a vital function in this regard because they have the ability to require that patient safety be an explicit criterion for certification of EHRs. Doing so would be an important first step.
Another step that can be taken by the private sector prior to implementation of health IT is for vendors and manufacturers to declare they have addressed safety issues in the design and development of their products, both self-identified issues and those detected during product testing. Such a declaration ought to include the safety issues considered and the steps taken to address those issues. A similar declaration for usability has been supported by the National Institute of Standards and Technology (NIST), which has developed a common industry format for health IT manufacturers to declare that they have tested their products for usability and asks manufacturers to show evidence of usability (NIST, 2010). Declarations also ought to be made with respect to vendor tests of a health IT product’s reliability and response time, both in vitro and in situ (Sittig and Classen, 2010). Such declarations could provide users and purchasers of health IT with information as they determine which products to acquire. Additionally, vendors can help mitigate safety risks by employing high- quality software engineering principles, as discussed in Chapter 4.
Usability represents an exceptionally important issue overall, and undoubtedly affects safety. However, it would be challenging to mandate usability. Although some efforts are beginning to develop usability standards and tools as discussed in Chapter 4, more publicly available data about and testing regarding usability would be helpful in this area. EHRs should increasingly use standards and conformance testing to ensure that data from EHRs meet certain standards and would be readable by other systems to enable interoperability is practical.
Besides providing feedback to vendors about their products, users also have important responsibilities for safety during the preimplementa- tion phase. Users need to make the often difficult and nuanced decision of choosing a product to purchase, as discussed in Chapter 4. If a product does not meet the needs of the organization and does not appropriately
interface with other IT products of the organization, safety problems can arise. Similarly, organizations need to be ready to adopt a new product in order for the transition to be successful.
Implementation
Industry-developed recommended (or “best”) practices and lessons learned ought to be shared. There are instances where generic lessons are learned and recommended practices can be shared between health professionals through mediums such as forums, chat rooms, and conferences. Lessons can also be shared through training opportunities such as continuing professional development activities. Questions arise such as whether to roll out a health IT product throughout an entire health care organization at once or in parts. Health care providers are continually attempting to determine the most effective configuration of health IT products for their own specific situations (e.g., drug interactions should be displayed as warnings in such a way that clinicians do not suffer from alert fatigue, leading clinicians to turn off all alerts). A user’s guide to acquisition and implementation ought to be developed by both the private and public sectors. Some efforts are currently under way, including programs at HIMSS and the ONC, but more work is needed, and the committee believes that such user’s guides should receive public support, though they might be developed by private entities.
Opportunities also exist for users to learn more about safer implementation and customization of health IT products. For example, what lessons have been learned regarding customization of specific health IT products? What experiences have others had integrating a specific pharmacy system with a particular EHR? Lessons from such experiences, once they are widely shared, can greatly impact implementation. It will be critical for users and vendors to communicate as health IT products are being implemented to ensure they are functioning correctly and are fitting into clinical workflow.
Postimplementation
Similar to the preimplementation and implementation phases, standards and criteria will be necessary to ensure that users have appropriately implemented health IT products and integrated them into the entire socio- technical system. In the postimplementation phase, the largest share of the work involves health professionals and organizations working with vendors to ensure patient safety.
Postimplementation tests, as discussed in Chapter 4, will be essential to monitoring the successful implementation of health IT products. Few tests currently exist, and more will need to be developed. For example, self-assessments
could monitor the product’s down time, review the ability to perform common actions (e.g., review recent lab results), and record patient safety events. Ongoing tests of how the product is operating with respect to the full sociotechnical system could identify areas for improvement to ensure the product fits into the clinical workflow safely and effectively (Classen and Bates, 2011; Sittig and Classen, 2010; Sittig and Singh, 2009). These tests are also a way for users to work with vendors to ensure that products have been installed correctly. Developers of these postimplementation tests should gather input from health care organizations, clinicians, vendors, and the general public. Similar to how organizations such as the Leapfrog Group validate tests for effective implementation of computerized provider order entry systems, an independent group ought to validate test results for implementation of all health IT. Conducting these tests is so important for ensuring safety that they ought to become a required standard and linked to a health care organization’s accreditation through The Joint Commission or others. Periodic inspections could also be conducted onsite by these external accreditation organizations (Sittig and Classen, 2010). Other ways to require these tests be performed include actions from the public sector, including regulation, such as including postimplementation testing in meaningful use criteria, but the committee feels postimplementation testing is too important to be tied only to the initiatives of a particular government program. These issues of safer implementation and safer use will continue to be an ongoing challenge with each new iteration of software and will continue to be an important area of focus long after the meaningful use program is completed.
Training Professionals to Use Health IT Safely
Education and training of the workforce is critical to the successful adoption of change. If the workforce is not educated and trained correctly, workers will be less likely to use health IT as effectively as their properly trained counterparts. Educating health professionals about health IT and safety can help them understand the complexities of health IT from the perspective of the sociotechnical system. This allows health professionals to transfer context- and product-specific skills, and therefore to be safer and more effective. For example, a team of clinicians using a new electronic pharmacy system needs to be trained on the functionalities of the specific technology. Otherwise, the team is susceptible either to being naive to the abilities of the technology or to unnecessarily developing workarounds that may undermine the larger sociotechnical system.
As discussed in earlier chapters, basic levels of competence, knowledge, and skill are needed to navigate the highly complex implementation of health IT. Because health IT exists at the intersection of multiple disciplines, a variety of professionals will need training in this relatively new
discipline that builds off the established fields of health systems, IT, and clinical care (Gardner et al., 2009). Training in the unique knowledge of health IT will be important, particularly for organizational leadership, IT professionals, and health professionals.
Organizational Leadership
Often it is the leadership of a health care organization that makes decisions about the various types of technologies to acquire based on the needs of the clinicians and patients. However, the decisions related to acquiring health IT are more complicated than acquiring other types of technologies such as computed tomography scanners and magnetic resonance imaging machines and require a specific body of knowledge. In order to select the most appropriate health IT product and make informed decisions, organizational leaders will need to be aware of specific considerations, such as whether to implement a health IT product all at once or in a sequential fashion, whether the vendor or the organization should be responsible for backing up data, and whether the health IT product will need to be interoperable with other products already in place, both internal and external to the organization. Understanding the issues related to health IT and its potential effects on patient safety is critical because of the large investment and continued resources needed for safer, more effective implementation of health IT.
The role of the chief medical and/or nursing information officer has been designed to be vital to the success of health IT, serving as a bridge between IT staff and clinicians. Small practices may relate to local hospitals or assign someone within their office, whether a clinician or not, to help lead these efforts. For example, the Chief Medical Information Officer Bootcamp offered by AMIA and tutorials or program offerings by organizations such as AMIA, the American Medical Directors of Information Systems, the College of Healthcare Information Management Executives, and the Scottsdale Institute can help identify and train appropriate personnel. Additionally, a better connection between the clinical informatics community and medical and nursing specialty society organizations would be helpful, such as the chief nursing information officers link promoted by HIMSS. Appointing a chief medical and/or nursing information officer allows for health care organizations to hold someone accountable for the implementation and use of health IT. As discussed in Chapter 3, truly being able to use health IT products and ensuring their safety requires a unique set of skills.
IT Professionals
With respect to the implementation and maintenance of technologies, a growing number of IT professionals are being trained to work with clinicians to redesign workflow as well as to manage and support implementation of health IT. The ONC supports a workforce development program to provide training at these levels. In May 2011, the ONC released a series of health IT competency exams for individual health IT proiessionals to demonstrate their knowledge level in acquiring health IT products, implementing and maintaining them, and training other staff on how to use them (ONC, 2011). IT professionals provide a key support function in health IT-enabled care delivery and therefore need to be trained on clinical workflows to best support clinicians and understand why workarounds that can lead to unsafe care are developed.
Health Professionals
While health professionals are rarely specialists in technologies themselves, they bear arguably the greatest responsibility for daily use of the technology and likely feel substantial responsibility to ensure that health IT products are not harmful to their patients. To optimize the potential for health IT to improve the safety of patient care, health professionals must not only learn how to use specific health IT products and about their full functionality but also learn how to incorporate these products into their daily workflows. If not properly trained on how to use a specific technology, health professionals may not only miss an opportunity to make their own processes safer and more efficient, but also may in fact develop unsafe conditions. At a minimum, clinicians need to be trained to recognize that health IT can improve quality of care while being cognizant of its potential to negatively impact patient safety if used inappropriately.
To date, programs focusing on training health professionals to use health IT generally are not widespread. As part of the ONC’s health IT workforce development program, health professionals can learn techniques related to implementation. Although this is a step in the right direction, other, more specific and comprehensive programs are needed to complement this training that focus more specifically on using health IT and to do so in an interprofessional manner. The opportunity to provide care in interprofessional teams and improve communications may be essential to providing safer care, shifting the paradigm of health care delivery. In addition to the fact that almost all health IT products are configured differently, health professionals’ interaction with IT products differs by specialty, by profession, by health care setting, and even by state.
Clinician education and training can be encouraged through a number
of avenues, including formal education and postgraduate training as well as the longer course of clinicians’ careers. Introducing concepts of health IT safety early in professional clinical training (e.g., professional school, residencies) allows clinicians the opportunity to learn how to use and practice delivery of care safely and effectively with a technology in place. As future generations of health professionals grow up using these technologies, they will be more adept at using them on a daily basis throughout their careers. It is also important to be trained in a local context (e.g., by hospital, clinic, nursing home). AMIA has had a program identified as “10x10” to offer rigorous graduate training-level introductory courses in clinical informatics, of which safety is generally a part. Further dissemination and offering of these courses could result in important advances.
AMIA, in partnership with the Robert Wood Johnson Foundation, has also developed a clinical informatics subspecialty through the American Board of Medical Specialties. Many boards are beginning to require a facility with health IT as part of a clinician’s maintenance of certification. Some specialty societies such as the American College of Physicians, the American Academy of Pediatrics, and the American College of Surgeons have active informatics committees that could explicitly take up the issue of safety, for discussions relating to both problems and solutions as well as creating a wider array of educational offerings.
To varying degrees, hospitals and other health care organizations require health professionals to be able to use health IT. For example, they often require clinicians to receive a certain number of hours of introduction to the health IT product before granting privileges.
Health IT can facilitate communication in team-based care. As a result, a focus on health IT safety is needed in interdisciplinary settings. An interdisciplinary focus on health IT safety that includes a variety of professionals is essential to safer, widespread use of health IT. A number of organizations are currently focusing on interdisciplinary team training. Nursing groups such as the Alliance for Nursing Informatics, the American Association of Colleges of Nursing, the National League for Nursing, and the American Organization of Nurse Executives are engaging in informatics and ought to continue developing concerted efforts to include safety issues in nursing programs. In addition, the Quality and Safety Education for Nursing program funded by the Robert Wood Johnson Foundation has developed a nursing curriculum that includes pathways for developing knowledge, skills, and attitudes related to patient safety and informatics (Quality and Safety Education for Nurses, 2011). Similar programs focusing on pharmacy training are also under way. For example, the American Society for Health-System Pharmacists has a membership section for pharmacy informatics and technology that sponsors educational programs at their annual meetings. An increasing number of institutions have postgraduate
pharmacy informatics specialty residency programs. The leading national pharmacy societies have formed the Pharmacy e-Health Information Technology Collaborative, for which training is a foundational goal. Other health professionals such as physician assistants and physical therapists also need training on health IT, some of which already have efforts under way. Health care executives through organizations such as the American College of Physician Executives also ought to be involved in these efforts.
Most of the foregoing comments relate to education more than simply training, especially with regard to the proper use of specific EHR products and systems. However, training is just as critical to safe use as education and is necessary for users to become aware of limitations of the software and also potential problems that have arisen elsewhere. Hospitals typically have training programs and labs available for clinicians to “dry run” the product and dedicated staff on wards or in clinics to serve as resources for others. Training sessions may be few or many. With specific attention to safety, staff need to know how the software deals with updates, for example drug-drug interactions. Greater attention to the creation of training modules focused on safety is needed, particularly on the strengths and weaknesses of both the software and the institution with respect to its early use of the system.
The committee believes it is incumbent on all stakeholders to participate in the creation of shared learning environments. It is also important to realize that the goal is not only learning by individuals but equally, if not more importantly, learning by the system itself. Through shared learning, health IT products can adapt to new challenges and opportunities while becoming safer over time.
THE ROLE OF THE PUBLIC SECTOR: STRATEGIC GUIDANCE AND OVERSIGHT
The first four recommendations are intended to encourage the private sector to create an environment that facilitates understanding of the risk, gathers data, and promotes change. But in some instances, the private sector cannot create this environment itself. The government in some cases is the only body able to provide policy guidance and direction to complement, bolster, and support private-sector efforts and to correct misaligned market forces. While a bottom-up approach may yield some improvements in safety, it is limited in its breadth. Gaps will arise that require a more comprehensive approach, for example, ensuring processes are in place to report, investigate, and make recommendations to mitigate unsafe conditions associated with health IT-related incidents. This section addresses solutions to some of these gaps.
The Health Information Technology for Economic and Clinical Health
legislation has embarked the nation upon a $30 billion investment that will impact both technology and clinical practice on a scale heretofore unseen in the nation (Laflamme et al., 2010). However, it is unclear what value the nation is receiving for its investment. Thus, the public sector has a responsibility to monitor and assess its investment and ensure that it is not harmful to patients. The ONC, AHRQ, the CMS, the National Institutes of Health, the Food and Drug Administration (FDA), and NIST, among other federal agencies, are becoming more actively involved in guiding the future direction of health IT, indicating that patient safety will also require a shared responsibility among federal agencies. However, currently funded government programs are just one part of a multifaceted solution to improving patient safety and health IT.
An appropriate balance must be reached between government oversight or regulation and market innovation. As with most rapidly developing technologies, governmental involvement has the potential to both foster and stifle innovation (Grabowski and Vernon, 1977; Hauptman and Roberts, 1987; Walshe and Shortell, 2004). For example, blood banking has been regulated for some time, and many believe regulation has limited innovation in this domain (Gastineau, 2004; Kim, 2002; Schneider, 1996; Weeda and O’Flaherty, 1998). Stringent regulations, while intended to promote safety, can negatively impact the development of new technology (Grabowski and Vernon, 1977) by limiting implementation choices and restricting manufacturers’ flexibility to address complex issues (Cohen, 1979; Marcus, 1988) and may even adversely affect safety. However, regulations also have the potential to increase innovations (see Appendix D) and improve patient safety, especially if manufacturers are not adequately addressing specific safety-related issues.
The committee could not identify any definitive evidence about the impact regulation would have on the innovation of health IT. Because health IT is a rapidly developing technology and its impact on patient safety is highly dependent on implementation and design, the committee believes policy makers need to be cognizant of not restricting the positive innovation or flexibility needed to improve patient safety. However, legal barriers imposed by contracts between vendors and users may prevent the sharing of safety-related health IT information and hinder the development of a shared learning environment. To encourage innovation and shared learning environments, the committee adopted the following general principles for government oversight:
• Focus on shared learning,
• Maximize transparency,
• Be nonpunitive,
• Identify appropriate levels of accountability, and
• Minimize burden.
The committee considered the various levels of oversight that would be appropriate and focused on areas where private-sector efforts by themselves would not likely result in improved safety. In particular, the committee considered a number of mechanisms to complement the private sector in filling the previously identified knowledge gaps. To achieve safer health care, the committee considers the following actions—when coupled with private- sector efforts—to be the minimum levels of government oversight needed:
• Vendor registration and listing of health IT products;
• Consistent use of quality management principles in the design and use of products;
• Regular public reporting of health IT-related adverse events to encourage transparency; and
• Aggregation, analysis, and investigation of reports of health IT- related adverse events.
The committee categorized health IT-related adverse events as deaths, serious injuries, and unsafe conditions.4 While deaths and serious injuries are concrete evidence of harm that has already occurred, unsafe conditions can represent the precursors of the events that cause death and serious injuries and are generally greater in volume and provide the proactive opportunity to identify vulnerabilities and mitigate them before any patient is harmed. Analysis of unsafe conditions would produce important information that could potentially have a greater impact on systemically improving patient safety and would enable the adoption of corrective actions that could prevent death or serious injury.
Registration of Vendors and Listing of Products
As a first step toward transparency and improving safety, it is necessary to identify the products being used and to whom any communications or actions about a product need to be directed. As learned from a variety of industries (such as medical devices and pharmaceuticals), vendor registration and listing of products is necessary to hold makers accountable for the safety of their products. Registration and listing can also serve as a resource
![]()
4 The committee recognizes that a number of terms are used to describe potential events and conditions unsafe for patients, otherwise known in this section as unsafe conditions. Other terms include close calls, near misses, hazards, and malfunctions that could lead to death or serious injury.
for purchasers to know what products are available in the market when selecting a health IT product. Having a mechanism to accomplish this is important so that when new knowledge about safety or performance arises that is specific to a product and is developed independent of a vendor, other users, and products that could also be vulnerable, can be identified.
The committee believes it is important that vendors of complete EHRs and EHR modules5 register with a centralized body and continue to list their products. This should eventually include vendors of all health IT products. Seeing that creators of internally developed EHR systems are also considered under the meaningful use program, they should continue to be included as vendors that need to register and list their products.
In determining what organization would be appropriate to register manufacturers and house a list of products, the committee considered FDA and the ONC as the most logical organizations to support these functions. According to the director of FDA’s Center for Devices and Radiological Health, FDA has authority to regulate health IT but has not yet exercised it for some types of health IT such as EHRs6 (Shuren, 2010). FDA currently requires that manufacturers of pharmaceutical drugs and medical devices register with FDA and list their products through a fee-based system. However, pharmaceutical drugs and medical devices are very different from health IT products such as EHRs, health information exchanges, and personal health records.
As part of the meaningful use program, the ONC employed a similar mechanism for EHR vendors to list their products. For health care providers to be able to qualify for EHR incentive payments, they must use a complete EHR or EHR module that meets specific certification criteria established by the ONC. ONC-ATCBs certify specific products based on these criteria and report certified products to the ONC on a weekly basis. The ONC collects and makes this information available to the public; the data are also available to allow purchasers to make more informed decisions about the systems they are interested in purchasing.7 The ONC therefore already has relationships in place with vendors and would be able to conduct this function without requiring a new mechanism, limiting any confusion that may arise as a result of duplicating an already-existing system.
The committee concludes that the ONC is a better option than FDA
![]()
5 The ONC defines a complete EHR as a “technology that has been developed to meet, at a minimum, all applicable certification criteria adopted by the Secretary” and an EHR module as “any service, component, or combination thereof that can meet the requirements of at least one certification criterion adopted by the Secretary” (HHS, 2010).
6 FDA regulates some types of health IT, for example tools for storing laboratory data, decision support (e.g., prescription dose calculators), and technology related to blood banks (Shuren, 2010).
7 The certified health IT product list can be found at http://onc-chpl.force.com/ehrcert (accessed April 20, 2011).
and supports continuation of the ONC’s efforts to list all products certified for meaningful use in a single database as a first step for ensuring safety.
Recommendation 5: All health IT vendors should be required to publicly register and list their products with the ONC, initially beginning with EHRs certified for the meaningful use program.
Adopting Quality Systems Toward Safer Health IT
The committee also considered enhancing the mechanisms for health IT vendors to ensure the safety of their products. Quality management principles and processes have been in place in industries such as defense and manufacturing since the early 20th century. These quality management principles and processes were first developed in response to military needs for quality inspection and evolved into a more comprehensive approach of identifying responsibilities for the production staff (American Society for Quality, 2011). These industries’ adoption of formal processes to continually improve the effectiveness, efficiency, and overall quality of their systems helped drive their products toward safety and reliability while still supporting innovation.
The same outcome can occur in health IT. Because quality management principles and processes focus on driving performance characteristics at each level to make sure that the product and specifications are in line with the users’ needs and expectations, they can help health IT vendors take into account characteristics such as interoperability, usability, and human factors principles as they design and develop safer products.
Examples of Quality Management Principles and Processes
Numerous industries have set standards for quality management principles and processes to help identify, track, and monitor both known and unknown safety hazards. Some examples of quality management principles and processes include the following: the Hazard Analysis Critical Control Point (HACCP) system, FDA Quality System Regulation (QSR), and the International Organization for Standardization (ISO) standards and principles. These examples of quality management principles and processes are designed to afford organizations the flexibility to develop their own processes to best suit their needs as long as these processes meet standard criteria. The examples presented are not meant to be inclusive or to indicate that one process is superior to the other. They are instead meant to illustrate what currently exists in other industries, what could be included in a process to ensure safety and quality with health IT, and how such processes could benefit the health IT industry.
A HACCP system, primarily used by the food industry as a quality system, is a proactive and preventive approach to applying both technical and scientific principles to ensure the reliability, quality, and safety of a product (Dahiya et al., 2009). Prior to implementation of a HACCP system, certain organizational prerequisites and preliminary tasks must be conducted to ensure that the environment supports the production of safe products. These tasks include assembling a team of individuals knowledgeable about the product and the processes involved in creating the product, as well as describing the product, its intended use, and its intended distribution. After these tasks have been completed, the focus can shift to implementing a HACCP system.
HACCP systems comprise seven principles that work together to help identify, prevent, track, and monitor hazards and aid in reducing risks that may occur at specific points in the product life cycle (see Figure 6-1) (Dahiya et al., 2009). Several of these principles can be important for use within the health IT industry, including the following: conducting a hazard analysis, determining critical control points, establishing monitoring procedures, and establishing corrective actions. Conducting a hazard analysis allows unsafe conditions to be identified prior to product completion and implementation in a health care setting. This is a preventive measure that enables vendors to determine critical control points that are then applied to prevent, eliminate, or mitigate the risk. These critical control points are then monitored to determine if they are under control or if a deviation has occurred. Finally, corrective actions are established to help determine the cause of a product deviation and what actions if any were taken to correct the issue. HACCP systems are used in the dairy, meat, and fish and seafood industries and are currently being adapted for use in the pharmaceutical industry.
Another example of quality management principles and processes can be found in FDA’s QSR for medical devices, which are also called current good manufacturing practices. A QSR is a set of interrelated or interacting elements organizations use to direct and control how quality policies are implemented and how quality objectives are achieved. FDA’s QSR helps ensure medical devices are safe and effective for their intended use by requiring both domestic and foreign device manufacturers to have a quality system that accomplishes the following outcomes: establish various specifications and controls for devices; design devices to meet the specifications of a quality system; manufacture devices based on the principles of a quality system; ensure finished devices meet these specifications; correctly install, check, and service devices; analyze quality data to identify and correct quality problems; and process complaints (FDA, 2009).
Like the HACCP principles, the subsystems under the FDA quality management principles and processes (see Figure 6-2) may be applied to

FIGURE 6-1
Principles of the HACCP system.
SOURCE: Adapted from Dahiya et al. (2009).
health IT. In particular, the management subsystem and the production and process controls subsystem may be useful in considering what quality management principles apply to health IT. The management subsystem can be useful within health IT because it emphasizes the importance of management’s role in making sure quality management principles and processes
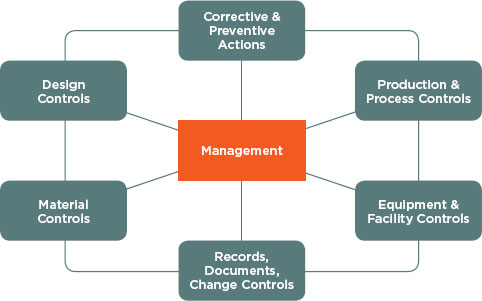
FIGURE 6-2
QMS subsystems.
SOURCE: FDA (2011).
are in place, such as completing quality audits to ensure employee compliance, and making employees aware of known product defects and errors. In addition, the production and process control subsystem allows for changes within product specification, methods, and procedures by allowing revalidation by the vendor to ensure that changes have not altered the intended use and usability of the product. This validation would be done by inspecting, testing, and verifying that the product conforms to its intended use and distribution.
Another proactive technique to characterize hazards, prioritize them for mitigation, and develop mitigation action plans is the Healthcare Failure Modes and Effects Analysis (HFMEA®). This methodology was developed by the National Center for Patient Safety at the U.S. Department of Veterans Affairs (VA) and is an amalgam of HACCP and traditional failure modes and effects analysis. HFMEA has been used for many patient safety- related issues including bar-coding for medication administration (DeRosier et al., 2002).
Another example of quality management principles and processes can be found with ISO, which sets standards for industries around the world. The ISO 9000 series is a quality management process based on eight principles to be used by an organization’s senior management as a framework toward improved performance (ISO, 2011). The eight principles
are customer focus, leadership, involvement of people, process approach, systems approach management, continual improvement, factual approach to decision making, and mutually beneficial supplier relationships. Generally, these principles are considered to be more comprehensive than other quality management principles and processes, including both HACCP and FDA QSR, because they apply basic quality controls to the whole system, including design and servicing. In addition to quality, ISO also provides guidance in the areas of safety and effectiveness and risk management. Finally, ISO standards have been developed to provide guidance on health informatics to manufacturers interested in producing EHRs as well as supporting interoperability and usability.
Application to Health IT Vendors
The adoption of quality management processes and principles have been demonstrated to improve product safety in other fields; therefore, the committee believes the adoption of quality management principles and processes to be critical to improving the safety of health IT (Chow-Chua et al., 2003; Glasgow et al., 2010, Johansen et al., 2011; VanRooyen et al., 1999). The committee is aware that many vendors already have some type of quality management principles and processes in place. However, not all vendors do, and the level of comprehensiveness of these quality management principles and processes is unknown. An industry standard is needed to ensure comprehensive quality management principles and processes are adopted throughout the health IT industry to provide health care organizations and the general public assurance that health IT products meet a minimum level of safety, reliability, and usability.
To this end, the committee believes adoption of quality management principles and processes should be mandatory for all health IT vendors. Oversight is needed to ensure compliance with these quality management principles and processes once they are in place. The committee considered four entities that could potentially oversee this process: FDA, the ONC, current ONC-ATCBs, and professional societies such as HIMSS.
As discussed earlier, vendors of FDA-regulated industries (e.g., medical devices, food, biologics, drugs) are required to have adopted quality management principles and processes. This risk-based framework is periodically inspected either when a concern is suspected or as part of a schedule and allows vendors to assess regulatory compliance. In addition, FDA has the ability to enforce and take other actions against those vendors who are not compliant with these regulations. Although the QSR provides vendors with flexibility, the committee does not believe the current quality system is the correct process needed for overseeing the health IT industry because it emphasizes
regulation of design and labeling. Instead, there may be principles within the QSR that may provide guidance to vendors of health IT.
The committee also considered the ONC’s potential role. Although the ONC is not operationally oriented like FDA, it already has relationships with health IT vendors. The ONC could build on the principles of the aforementioned examples and develop a standard baseline set of principles upon which vendors could conform their products and processes. This would create a minimum standard for the industry while also allowing vendors flexibility without fear of regulation.
Another option is for the ONC-ATCBs to require that such processes be adopted as part of their certification criteria for the meaningful use program. Vendors who wish to have their EHR certified for meaningful use are currently working with these certifying bodies, so a relationship already exists. While oversight would fall to these certifying bodies, they could coordinate with the appropriate organization to develop processes that would aid in guiding the development of safer health IT. However, having the responsibility diffused among a number of the ONC-ATCBs may be problematic because of the difficulty of ensuring all organizations hold the vendors to the same level of scrutiny. Furthermore, the committee considers quality management principles and processes to be critical for the safety of health IT products and therefore does not want to limit the adoption of quality management principles and processes to those vendors participating in the meaningful use program.
Finally, the committee discussed the role of professional associations and societies such as HIMSS. Professional associations can help to provide guidance to the industry by setting and adopting current national and international standards, establishing industry recommended practices, and issuing recommendations. Similar to the ONC-ATCB option, professional associations in turn could work with a governmental body like FDA or an organization like ISO to establish guidelines comparable to those in other industries. However, there is concern that HIMSS and other professional associations and societies may not be appropriate because they have a significant amount of industry sponsorship that could jeopardize their ability to serve as neutral parties.
The ONC, FDA, and the ONC-ATCBs are examples of organizations that could all potentially administer this function. In the absence of other information, the Secretary of HHS should determine the most appropriate body.
Recommendation 6: The Secretary of HHS should specify the quality and risk management process requirements that health IT vendors must adopt, with a particular focus on human factors, safety culture, and usability.
In addition to vendors, users of health IT ought to have quality management principles and processes in place to help identify risks. However, the committee could not find any documented evidence to suggest which processes would lead to better outcomes among users.
Regular Reporting of Health IT-Related Deaths, Serious Injuries, or Unsafe Conditions
As discussed previously, to ascertain the volume and types of patient safety risks and to strengthen the market, reports of adverse patient safety events need to be collected. Regular reporting of adverse events is a widely used practice to identify and rectify vulnerabilities that threaten safety. When adverse events are identified, organizations not only learn from their mistakes but can also share the lessons learned with others to prevent future occurrences. As described in Chapter 3, event reporting is critical to promoting safer systems, especially when work-as-designed drifts from work- in-practice. The previously discussed private-sector reporting system for identifying health IT-related adverse events, while critical, only encourages reporting to a certain level. To drive reporting, it needs to be supplemented with a comprehensive reporting system.
Reporting Systems in Other Industries
The experiences of reporting systems both in health care and other industries have shown that it is critical to create an environment that encourages reporting. In the United States, a number of industries have mechanisms to collect reports of adverse events, including aviation, rail, industrial chemicals, nuclear power, and pharmaceuticals. One leading example of a reporting system for close calls is the NASA Aviation Safety Reporting System (ASRS). Under the auspices of ASRS, reports of hazards in which aviation safety may have been compromised are submitted voluntarily (accidents—instances involving actual harm—are not accepted and are referred to other agencies such as the Federal Aviation Administration [FAA] and the National Transportation Safety Board [NTSB]). ASRS compiles the deidentified, verified reports in a publicly accessible database. Once the reports have been deidentified, NASA retains no information that would allow association with the initial reporter; the program has not had a breach in confidentiality in its more than 30-year history. The FAA also makes two other commitments to the aviation community: (a) it does not use ASRS reports against reporting parties in enforcement actions, and (b) it waives fines and penalties under many circumstances for unintentional
violations of federal aviation statutes and regulations that are reported to ASRS (NASA, 2011a).
Provision (a) states that information provided by reporting parties will not be used against them, and it addresses the fear that information contained in a report will be used against the reporting party. Provision (b) goes farther in that it offers actual immunity under specified circumstances for safety incidents if a report has been properly filed, regardless of how knowledge of the harm comes to light. (Reporting parties may be found in violation of an aviation regulation, but no punitive actions will be taken under specified circumstances.) Provision (b) thus provides an affirmative incentive for reporting (NASA, 2011b).
Despite the success of ASRS, it is not a comprehensive system. ASRS does not receive reports of actual adverse events and does not have the ability to conduct thorough investigations as to causative factors within the sociotechnical system. Therefore, confidentiality and immunity are only conferred for reports of close calls and not actual harm. Identifying patterns of occurrences is not the same as identifying the underlying causes. This is a critical point because without identification of the underlying causes, it can be difficult if not impossible to formulate a coherent and effective action plan to mitigate the risk to patients.
Reporting Systems in Health Care
Testimony to the U.S. Senate in 2000 noted that “[a]pproaches that focus on punishing individuals instead of changing systems provide strong incentives for people to report only those errors they cannot hide. Thus, a punitive approach shuts off the information that is needed to identify faulty systems and create safer ones. In a punitive system, no one learns from their mistakes” (U.S. Congress, 2000). Reporting systems currently exist in health care to protect patient safety both in other countries and within the United States. Reporting systems have long been in place in countries such as Denmark, the Netherlands, Switzerland, and the United Kingdom. These systems all differ with respect to their design, but some common themes have emerged: reporting ought to be nonpunitive, confidential, independent, evaluated by experts, timely, systems-oriented, and responsive. The term nonpunitive is used here to mean that reports of health IT-related adverse events should be free from punishment or retaliation as a result of reporting. Some systems mandate reports of adverse events and patient safety incidents, while others allow for confidential, voluntary reporting (WHO, 2005).
Within the United States, a few disparate adverse event reporting systems have been developed by groups such as The Joint Commission, the Department of Veterans Affairs (VA), individual states, and more recently at a national level, through the patient safety organizations (PSOs):
• The Joint Commission requires reports of sentinel events8 to be submitted for health care organizations to receive accreditation. Organizations are expected to conduct their own investigations of sentinel events and submit a report of both the process and results to The Joint Commission for verification. If reports are not up to standard, organizations have the opportunity to fix the reports or their accreditation status may be reviewed (The Joint Commission, 2011).
• The VA developed two complementary nonpunitive reporting systems, the external NASA/VA Patient Safety Reporting System and the internal VA National Center for Patient Safety reporting system. The NASA/VA Patient Safety Reporting System is an external, nonpunitive system modeled after NASA ASRS and accepts reports of close calls and actual adverse events where harm has occurred. Reports are protected from disclosure and no individuals are identified explicitly in these reports or in the subsequent safety investigations. Like its sister system, NASA ASRS, the NASA/VA Patient Safety Reporting System does not do comprehensive investigations as to causation or track implementation for countermeasures. It was developed as a safety valve where reporters unwilling to report to the internal VA National Center for Patient Safety reporting system could make their concerns known. The internal, nonpunitive VA National Center for Patient Safety reporting system received more than 1,000 reports for every report received by the NASA/VA Patient Safety Reporting System over a 10-year period (Bagian et al., 2001). The VA National Center for Patient Safety reporting system does formulate action plans for reports of events, tracking implementation, effectiveness of interventions, and mechanisms for dissemination, and for this reason is the mechanism that affords the greater potential to improve safety.
• At the state level, 26 states currently require mandatory reporting of adverse events and 1 state has a system in place for voluntary reporting (National Academy for State Health Policy, 2010).
• FDA requires manufacturers of pharmaceutical drugs, therapeutic biologic products, and medical devices to report adverse events to its MedWatch program. The general public and health care providers also are encouraged, although not required, to voluntarily submit reports of adverse events to FDA.
• Clinicians and health care organizations can confidentially report patient safety work product (not just adverse events related to health IT) to the PSOs, which operate under the purview of
![]()
8 The Joint Commission defines sentinel event as “an unexpected occurrence involving death or serious physical or psychological injury, or the risk thereof” (The Joint Commission, 2011).
AHRQ.9 The PSOs are intended to analyze patient safety reports to identify patterns and to propose measures to eliminate patient safety risks and hazards.
Despite the existence of these and other systems and many calls for change, reports of adverse events in the United States currently are not collected in a comprehensive manner. Furthermore, learning from these systems is limited because a multitude of different data is collected by each system, hampering any attempt to aggregate data between reporting systems. It is also the case that users are being asked to notify multiple reporting systems; the burden of multiple reporting systems can potentially discourage reporting. As previously discussed, the Common Formats effort is under way to streamline the types of data being collected, which serves as a first step to collecting similar data across reporting systems. It must be emphasized, however, that these reporting systems are not intended or capable of furnishing accurate counts or prevalence and incidence data. Instead, their purpose is to identify vulnerabilities and hazards that can then be prioritized for the institution of corrective actions to mitigate the identified risks.
The committee believes systems that encourage both vendors and users to report health IT-related adverse events are needed to improve the safety of patient care. Evaluations of reporting systems examined by the committee indicate that well-designed voluntary reporting can improve safety (Leape, 2002) in an environment that encourages reporting. To create an environment that encourages reporting, reporters’ identities need to be kept confidential; it is important to note that protections of privilege and confidentiality generally refer to reports by users and not manufacturers of products. In addition, if the reports do not result in action or if actions that are taken are not provided as feedback to the reporters, the sustainability of the reporting system can be impacted negatively. Perhaps more important is that reports for the purpose of learning have to be separate from the purpose of addressing accountability. An environment that does not allow reporting parties to be punished as a result of reporting is critical for the success of voluntary reporting systems.
![]()
9 As of August 2011, 81 PSOs were listed (AHRQ, 2011b). These organizations receive reports of events from providers, provide feedback and recommendations to the reporting providers, and deidentify reports and send the data to AHRQ for incorporation into a network of patient safety databases. However, it is important that data received by the PSOs are not deidentified and scrubbed of all data that would make data unuseful for drawing trends, such as what technology, product, and module number are being used.
Reporting of Health IT-Related Adverse Events
A subset of adverse events can be identified as being related to health IT. Health IT-related events can occur in every setting and level of health care but currently are not required to be reported. As a result, shared knowledge of such events is incomplete and any effort to understand and prevent such events from occurring in the future is far from optimal. The committee believes it is in the best interest of the public for deidentified, verified health IT-related adverse events to be released transparently to the public for the purpose of shared learning, and the responsibility for identifying and reporting events lies with both vendors and users of health IT. Although reporting could also be done by patients and the general public, the committee believes vendors and users are the only actors that should be responsible formally for reporting adverse events and that they have distinct roles in this regard. Coupled with the suggested private-sector system for reporting, a comprehensive public-private system that supports an environment for reporting can be created.
Reports of health IT-related death, serious injury, and unsafe conditions should be collected. Those events falling into the unsafe conditions category may be especially difficult to detect, for example, events that arise as a result of usability issues that do not result in immediate patient harm. Based on the experiences from the VA National Center for Patient Safety reporting system and others, the committee concludes reporting systems should not receive submissions of intentionally unsafe acts;10 instead, intentionally unsafe acts ought to be relayed to the health care organizations themselves to follow regular legal channels. In this way, the goal of the reporting system remains focused on learning and not accountability.
Reports by Vendors
For health IT-related adverse events, the onus of reporting does not fall solely on users. The committee believes all vendors should collect and act on reports of adverse events. Some vendors collect, review, and act on reports of adverse events from both internal and external sources in a variety of ways (e.g., issue alerts to health care providers who may be affected) (IOM, 2011). Vendors are not required to report adverse events if their products are not regulated by FDA currently, although some health IT vendors voluntarily report to the FDA MedWatch Safety Information and Adverse Event Reporting Program (IOM, 2011). However, because vendors do not perform these tasks in a consistent way or share experiences,
![]()
10 Intentionally unsafe acts in this context can be defined as “events that result from a criminal act, a purposefully unsafe act, or an act related to alcohol or substance abuse or patient abuse” (Veterans Affairs National Center for Patient Safety, 2011).
instances of harm and any lessons learned from them are not shared systematically.
The committee concludes that reporting of deaths, serious injuries, or unsafe conditions should be mandatory for vendors and that an entity be given the authority to act on these reports. The ability to take action would allow imminent threats to safety to be addressed immediately and could provide valuable information for future activities and development work. These reports should be collected by an entity for the purpose of learning and therefore should not be used for punitive purposes. A mandate entails expectations that vendors will report adverse events and that some sort of penalty would result for failing to report events.
To create a program of mandatory reporting, direction will need to come from a federal entity with adequate expertise, capacity, and authority to act. This entity could either collect the reports itself or could coordinate with and delegate to the private sector to the extent possible. Precedence for delegation exists, such as the delegation of certification of EHRs by the ONC to a number of the ONC-ATCBs. FDA currently has the capability to require mandatory reporting if it exercised its discretion in regulating EHRs and it has an existing infrastructure for reporting adverse events, but, as discussed in previous sections, the committee does not believe FDA currently has the capacity to do so unless given adequate resources. An authority needs to be designated by the Secretary of HHS to require and act on reports of health IT-related adverse events, and be provided with resources to do so.
Reports by Users
Given the variability in implementing health IT, users of health IT— primarily clinicians as the end users and provider organizations—should be encouraged to report adverse events voluntarily, which should be non- punitive. One mechanism to encourage reporting is to have mechanisms available to easily report hazards and harms (e.g., a button embedded in an EHR screen that allows for one-click reporting). As stated earlier, self- reports are intended to identify vulnerabilities and hazards; they cannot be considered to represent incidence and prevalence of adverse events accurately or prove causation. This is illustrated by a recent study that found adverse events occur in 33 percent of hospital admissions but that local hospital reporting systems identified only 1 percent of those events (Classen et al., 2011), signaling underreporting in voluntary systems. Additionally, the numerous, uncoordinated reporting efforts can result in confusion as to what information ought to be reported, to what system, and in what format. A more streamlined system for reporting is needed so that people are not being asked to relay similar data about the same event multiple
times. The committee believes the complexity and frequency of reporting would make mandatory reporting for users infeasible. A reporting system that collects the same information precisely defining the components of a health IT-related adverse event is needed, such as the Common Formats.
The committee therefore concludes that reports by users should remain voluntary and that the identities of reporters should not be discoverable under any circumstance. User-reported health IT-related adverse events should be collected by a central repository and also be sent to the appropriate vendor. In this way, the vendor can also be notified of the event and take precautions and warn other users that may be at risk for an adverse event.
FDA, NASA, AHRQ, states, and others are all able to collect users’ reports of health IT-related adverse events. As discussed earlier, the complexity of health IT differs so greatly from FDA’s current portfolio that the committee believes FDA would have to institute approaches that are different from their current approach were they to oversee a health IT-related adverse event reporting program for users. NASA has operated ASRS and patient safety reporting systems and has a strong history in keeping reports confidential, but it is not clear that it is positioned to take on a responsibility of this magnitude. The PSO program is just beginning to take shape and is yet to be proven as a comprehensive network for report collection, but it boasts strong confidentiality protections and a widespread presence, and it is tied to a national patient safety database network. Many states have their own reporting requirements, but promoting a disjointed system of reporting limits the ability for systematic collection of reports across the country. The protections offered in the PSO program lead the committee to believe that the PSO program may be the best option for collecting reports from users, but further evidence will need to be collected to validate the success of this approach for user reporting of health IT issues.
Recommendation 7: The Secretary of HHS should establish a mechanism for both vendors and users to report health IT-related deaths, serious injuries, or unsafe conditions.
a. Reporting of health IT-related adverse events should be mandatory for vendors.
b. Reporting of health IT-related adverse events by users should be voluntary, confidential, and nonpunitive.
c. Efforts to encourage reporting should be developed, such as removing the perceptual, cultural, contractual, legal, and logistical barriers to reporting.
Perceptual, Cultural, and Logistical Barriers to Reporting
As mentioned earlier, multiple barriers prevent sharing and reporting of adverse events. In addition to the previously discussed legal issues, there are also perceptual, cultural, and logistical barriers to reporting. These barriers inhibit not only users’ abilities to report to the government, but also sharing of health IT-related adverse events with vendors, other users, and consumer groups. To have an adequate system of reporting and shared learning, each of these barriers needs to be addressed. Indeed, even the best- designed health IT product will inevitably have some content that might contribute to adverse events. Once this is recognized by both providers and manufacturers, disclosure of events can be viewed as the beginning of a shared effort to recognize, fix, and improve patient safety issues.
Clinicians may also be faced with cultural barriers within their own organization. Although reporting of all adverse events has been encouraged for many years, providers may be more reluctant to report those related to health IT because acquiring a health IT product is an expensive, lengthy, and time-consuming investment. After spending a lot of time and resources to adopt health IT, health care organizations may be reluctant to acknowledge that the product may be flawed. Therefore, in the eyes of management, reporting can be viewed as blaming decision makers for implementing a flawed system. This may lead to a culture where health professionals fear that reporting may be viewed as opposing management decisions. These organizations need to recognize that, no matter how well planned the selection and integration of health IT are, problems can arise. Organizational leadership needs to not only encourage reporting, but also view it as a necessary step to improve a complex system that will inevitably have problems.
The logistics of reporting may also be a barrier to reporting. Many health IT-related adverse events occur during treatment, when finding ways to continue care is a higher priority than reporting the problem. Afterward, clinicians may have little time to submit a report of the event or may not remember the full details of the event. Vendors need to create an easy system where clinicians can quickly and easily report. Also, clinicians may be less motivated to report when they do not believe vendors will use the report to change their systems. Vendors and users need to communicate more effectively by providing feedback to each other and making it known that reporting can lead to changes. Each organization and vendor will have its own unique set of barriers; however, the committee believes the potential benefits are worth the burden reporting would impose on vendors and users.
Aggregating, Analyzing, and Investigating Reports of Health IT-Related Adverse Events
The previous sections and recommendations detail a comprehensive plan that creates an environment to understand the risks and encourage change. This next section focuses on actions that can be taken in response to reports of health IT-related incidents to inform and prevent future events and unsafe conditions from occurring.
Reports of patient safety incidents are critical but are only one part of a larger solution to maximize the safety of health IT-assisted care. As discussed above, reports from vendors, users, or patients alone are generally not complete or detailed enough to understand fully the events leading to an incident and/or to develop solutions that would avert future problems. What is reported is often not completely reflective of what actually occurred.
As illustrated by the creation of the NTSB and the Nuclear Regulatory Commission (NRC) to oversee safety in high-reliability industries such as transportation and nuclear energy, the committee notes that the power to improve safety lies not only with requiring reports of safety incidents, but also with the ability to act on and learn from reports.
Thus, in addition to reporting, two additional activities are needed: (1) aggregating and analyzing data from reports and (2) investigating the circumstances and environment associated with safety incidents to try to identify the conditions that contribute to those incidents. Through these processes, lessons learned can be developed and disseminated so that similar incidents will be less likely to occur in the future. To maximize the effectiveness of reports, the collection, aggregation and analysis, and investigation of reports should be coupled as closely as possible.
Aggregation of reports of health IT-related adverse events—reports of deaths, serious injury, or unsafe conditions—should be conducted to identify patterns. Analyses of these reports can lead to identification of specific patient safety risks, potentially particular to a specific vendor or product. Such examination and additional investigation, where indicated, can lead to development of recommended practices or lessons learned and, potentially, prevent harm to patients.
Reports of adverse events can generate material that supports learning, but analysis is needed to obtain full value from such material. Although retrospective, an investigative function that comprehensively examines the conditions that contribute to health IT-related adverse events is critical so that concrete actions can then be taken proactively to reduce harm.
A Reporting System for Learning and Improving Patient Safety
On their own, vendors and users generally do not have the broad expertise needed to conduct investigations as envisioned by the committee. Vendors and users also may not be impartial arbiters of why an adverse event occurred. As a result, external methods are needed to conduct rigorous investigations of health IT-related adverse events.
Reports could be aggregated and analyzed by multiple entities, such as the PSOs, but trends in data may not be as easily identified if spread out among the more than 80 PSOs because of the smaller number of reports each organization would receive. Additionally, standards of analysis may not be used in the same way across multiple entities, calling into question the reliability of the analyses conducted. Ideally, as depicted in Figure 6-3, reports of health IT-related adverse events or unsafe conditions from both users and vendors would be aggregated and analyzed by a single entity that would identify reports for immediate investigation.
Reports to this entity would have to include identifiable data to allow for investigators to follow up in the event the reported incident requires investigation, but, as discussed previously, full confidentiality protections must be applied to the reports. Reports would need to be received in an identifiable manner from the PSOs or another collecting agency and with enough information to investigate (e.g., specific vendor, model number).
The entity would have the discretion to investigate two categories of reports: (1) novel reports that result in death or serious injury and (2) trends of reports of unsafe conditions (e.g., multiple health care organizations find that a specific pharmacy system accepts only 100 characters of a particular EHR’s notes section that allows 125 characters, resulting in the incorrect filling of orders). Prioritization among the reports would be determined on a risk-based hazard analysis. Cases resulting in death or serious injury should be investigated immediately. Reports should be kept confidential and nonpunitive for the purposes of learning. Reports of unsafe conditions should be analyzed and monitored continually and investigated using a risk- based hazard analysis. Which reports to investigate ought to be determined by the explicit risk-based prioritization system that the investigatory entity employs. Reports by vendors should already contain identifiable data.
In keeping with the principle of transparency, reports and results of investigations should be made public. Public release of results of investigations could build off the NTSB process, which separates facts discovered by the investigators from opinions and conclusions drawn by the investigators. A feedback loop from the investigatory body back to both the vendors and users is essential to allow groups to rectify any systemic issues that were found to introduce risk into their systems.
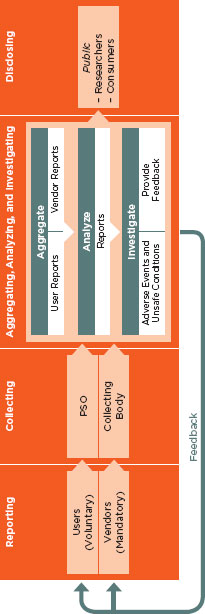
FIGURE 6-3
Reporting system for learning and improving patient safety.
Potential Actors
It is the intent of the committee to build on the current patient safety environment and to maximize the potential for all stakeholders to have a part in ensuring patient safety. However, for the reasons mentioned above, the current system does not adequately address the significant needs of a comprehensive process to learn from health IT-related patient safety risks. A unique knowledge base is needed to understand thoroughly and diagnose ways to improve the interface between health care delivery and health IT, which, as discussed in Chapter 3, is extraordinarily complex and requires the understanding of a large number of sociotechnical domains.
The committee considered a variety of alternatives to objectively analyze reports of unsafe events as well as conduct investigations into health IT-related adverse events in the way the committee envisions. The alternatives considered include FDA, AHRQ, the CMS, the ONC, and entities in the private sector.
• FDA is largely an oversight entity. Adding investigative responsibilities of the nature envisioned by the committee would be at odds with its oversight functions. Additionally, FDA lacks the resources in terms of capacity and expertise, limiting its ability to act in this area.
• AHRQ primarily supports research and technical assistance activities regarding quality and safety. It is not an oversight or investigative agency. AHRQ is also not an active implementer and is not operationally oriented. For AHRQ to take on the functions envisioned by the committee would require a complete change in the agency’s charge and internal expertise.
• The CMS is an administrative agency that has demonstrated its leverage with users of health IT through the development of the EHR incentive program and conditions of participation. These programs, while important, contain punitive elements and are inappropriate for completing the aggregative, analytic, and investigative functions described above. The CMS also has little internal expertise relating to clinical workflow and use of technology to support cognition and therefore does not have the infrastructure needed to support these tasks. Additionally, while the CMS could potentially serve in this role, the CMS’s authority focuses on providers associated with Medicare and Medicaid and is not as expansive as the committee believes is needed.
• The ONC coordinates health IT efforts and influences the development of policy related to health IT but has no formal authority in this area; it is not an operating division of HHS. It is not clear to
the committee that the ONC has the clinical and operations expertise to conduct investigations of health IT-related adverse events.
• Private-sector organizations such as The Joint Commission, the NCQA, and the NQF play an important role in ensuring safety. However, these organizations are mostly dependent on short-term funding streams, often seeking “soft money” to sustain specific projects and programs. As a result, programmatic content may be shaped in part by funders. Moreover, these groups—even working in collaboration with one another—may not have the far-reaching influences of a federal entity and do not have the mandate or breadth to be able to conduct such investigations at a national level. These groups also do not possess the expertise and ability to properly investigate these issues.
Investigating patient safety incidents related to health IT does not match the internal expertise of any existing entity. Given the status quo, the committee concludes that no existing entity has the necessary attributes to perform the crucial function of investigating health IT-related patient safety incidents as envisioned by the committee. The needed functions are under the jurisdiction of multiple federal agencies, and efforts are uncoordinated and are not as comprehensive as a safety-oriented system ought to be. A multiagency structure could be envisioned, but as discussed previously, oversight and investigative functions should not be housed in the same entity. The committee concludes the envisioned necessary functions cannot be realized solely through current structures, and a new entity is needed that can pull together the following desired goals in an integrated fashion in a way current alternatives cannot achieve:
• A comprehensive system for identifying and investigating deaths, injuries, and unsafe conditions has to be separated explicitly from oversight functions.
• Broad categories of expertise are required to investigate an adverse event fully.
• Vendors and users need to be held accountable publicly for patient deaths, injuries, and potentially unsafe conditions.
• A streamlined approach is needed to reduce wasteful duplication and inconsistencies.
• Lessons from these investigations need to be shared broadly from a respected source so that future adverse events can be averted.
To truly improve patient safety, a new approach is needed. The committee believed that the experiences of other industries such as transportation and nuclear energy in creating the NTSB and the NRC were instructive, and
concluded that the development of an independent, federal entity was best suited to performing the needed above-described analytic and investigative functions for health IT-related adverse events in a transparent, nonpunitive manner.
The committee envisions an entity that would be similar in structure to the NTSB or the NRC, which are both independent federal agencies created by and reporting directly to Congress. Among other responsibilities, these entities conduct investigations, for the purpose of ensuring safety. The NTSB is a nonregulatory agency that does not establish fault or liability in the legal sense but investigates incidents. The NRC is a regulatory body that has the ability to issue fines and fees. The committee considered both agencies and concluded the NTSB to be most similar to the needs of health IT-assisted care.
An independent, federal entity analogous in form and function to the NTSB is needed. This entity would not have enforcement power and would be nonpunitive. Instead, it would have the authority to conduct investigations and, upon their completion, make recommendations. The NTSB makes nonbinding recommendations to the Secretary of the Department of Transportation, who then must state within 90 days whether the department intends to perform the recommended procedures in total, carry the recommendations out in part, or refuse to adopt the recommendation. In this case, an entity would make similar recommendations to the Secretary of HHS. Although delivering nonbinding recommendations can be described by some as a flaw, the committee believes that the flexibility it provides is a strength, allowing for the health care organizations, vendors, and external experts to determine the best course forward collectively. If requested by the Secretary, the entity could also perform other functions, such as coordinating with existing bodies, both public and private, as appropriate. Investigations could involve representatives from all impacted parties, including vendors and users involved in the incident, as well as experts in the various sociotechnical dimensions of health IT safety. The committee believes that an independent, federal entity is the best option to provide a platform to support shared learning at a national level. The entity would have the following functions:
• Aggregate reports of health IT-related adverse events from at least vendors and users;
• Analyze the aggregated reports to identify patterns;
• Investigate reports of health IT-related patient deaths or serious injury;
• Investigate trends of reports of unsafe conditions;
• Recommend corrective actions to the Secretary of Health and Human Services;
• Provide feedback to vendors and users following investigations; and
• Disclose results of the investigations to the public, including researchers and consumers.
Recommendation 8: The Secretary of HHS should recommend that Congress establish an independent federal entity for investigating patient safety deaths, serious injuries, or potentially unsafe conditions associated with health IT. This entity should also monitor and analyze data and publicly report results of these activities.
It is also important to recognize that the line between health IT-related adverse events and other adverse events will likely become increasingly blurry. Multiple factors contribute to unsafe conditions and adverse events, making it potentially difficult to differentiate between health IT-related or other factors until an investigation has been conducted. If a broader system for all adverse events is created, the spirit of the committee’s recommendations should be recognized and considered.
Patients must be kept safe in the midst of the current large-scale rollout of health IT. While it is clear to the committee that the market has failed to keep patients safe with respect to health IT, the committee believes transparency is the key to improving safety. To truly address health IT safety, many actions will be needed to correct the market; ways to improve flow of communication and correct the market have to be created carefully. When combined, removing contractual restrictions, establishing public reporting, and having a system in place for independent investigations can be a powerful force for improving patient safety.
Achieving transparency, safer health IT products, and safer use of health IT will require the cooperation of all stakeholders. Without more information about the magnitude and types of harm, the committee concluded that other mechanisms were necessary to understand how to best approach health IT safety.
The committee believes the current state of safety and health IT is not acceptable; specific actions are required to improve the safety of health IT. The first eight recommendations are intended to create conditions and incentives to encourage substantial industry-driven change without formal regulation. However, because the private sector to date has not taken substantive action on its own, the committee believes a follow-up recommendation is needed to regulate health IT formally if the actions recommended
to the private and public sectors are not effective.11 If the first eight recommendations are determined by the Secretary of HHS to be not effective, the Secretary should direct FDA to exercise all authorities to regulate health IT.
The committee was of mixed opinion on how FDA regulation would impact the pace of innovation but identified several areas of concern regarding immediate FDA regulation. The current FDA framework is oriented toward conventional, out-of-the-box, turnkey devices. However, health IT has multiple different characteristics, suggesting that a more flexible regulatory framework will be needed in this area to achieve the goals of product quality and safety without unduly constraining market innovation. For example, as a software-based product, health IT has a very different product life cycle than conventional technologies. These products exhibit great diversity in features, functions, and scope of intended and actual use, which tend to evolve over the life of the product. Taking a phased, risk-based approach can help address this concern. FDA has chosen to not exercise regulatory authority as discussed previously, and controversy exists over whether some health IT products such as EHRs should be considered medical devices. If the Secretary deems it necessary to regulate EHRs and other health IT products not currently regulated by FDA, clear determinations will need to be made about whether all health IT products classify as medical devices for the purposes of regulation. The committee also believes that if FDA regulation is deemed necessary, FDA will need to commit sufficient resources and add capacity and expertise to be effective.
The ONC and the Secretary should examine progress critically toward achieving safety and, if needed, determine when to move to the next stage. HHS should report annually to Congress and the public on the progress of efforts to improve the safety of health IT beginning 12 months from the release of this report. In these reports, the Secretary should make clear why she does or does not believe further oversight actions are needed. In parallel, the Secretary should ask FDA to begin planning the framework needed for potential regulation consistent with Recommendations 1 through 8 so that, if she deems FDA regulation to be necessary, the agency will be ready to act, allowing for the protection of patient safety without further delay. FDA will need to coordinate these efforts with the actors identified in Recommendations 1 through 8, including AHRQ and the ONC, among others. In addition, the Secretary will also need to devise new strategies to stimulate the private sector to meet its responsibility of ensuring patient safety. The committee recognizes that not all of its recommendations can be acted on by the Secretary alone and that some will require congressional action.
![]()
11 One member disagrees with the committee and would immediately regulate health IT as a Class III medical device, as outlined in Appendix E.
Recommendation 9a: The Secretary of HHS should monitor and publicly report on the progress of health IT safety annually beginning in 2012. If progress toward safety and reliability is not sufficient as determined by the Secretary, the Secretary should direct FDA to exercise all available authorities to regulate EHRs, health information exchanges, and PHRs.
Recommendation 9b: The Secretary should immediately direct FDA to begin developing the necessary framework for regulation. Such a framework should be in place if and when the Secretary decides the state of health IT safety requires FDA regulation as stipulated in Recommendation 9a above.
Today the nation is just scaling up with EHRs, and, as a result, the health IT environment is both very dynamic and also stressed. Patient safety is too important to ignore, but clear routes to solid policies that will improve performance are still wanting. Because of the lack of concrete evidence about how to best improve patient safety, the private and public sectors should work together to take the first steps toward identifying the data and building an evidence base for improving health IT-related patient safety. Lessons should be learned from other industries focusing on safety. Although it is important to recognize that none of those reporting systems is perfect, a critical lesson to be learned from these experiences is that safety demands systems of continual learning.
Health IT is a quickly changing field, particularly with respect to outpatient services, and products are being developed continually for the improvement of patient outcomes and more effective care delivery. The functions and types of health IT-related adverse events requiring analysis and investigation will change over time. To this end, the approaches identified in this report should be monitored continually and revised as needed. The identified actor or set of actors should be given the flexibility and latitude to amend its charge as appropriate.
Creating an infrastructure that supports shared learning about and improving the safety of health IT is needed to achieve better health care. Proactive measures have to be taken to ensure health IT products are developed and implemented with safety as a primary focus through the development of industry-wide measures, standards, and criteria for safety. Surveillance mechanisms will be available to identify, capture, and investigate adverse events to improve the safety of health IT continually. Transparency and cooperation between the private and public sectors is the key to creating the necessary infrastructure to build safer systems that will lead to better care for all Americans.
AHRQ (Agency for Healthcare Research and Quality). 2011a. Common formats. http://www.pso.ahrq.gov/formats/commonfmt.htm (accessed May 8, 2011).
AHRQ. 2011b. Georgraphic directory of listed patient safety organizations. http://www.pso.ahrq.gov/listing/geolist.htm (accessed August 9, 2011).
American Society for Quality. 2011. The history of quality—overview. http://asq.org/learn-about-quality/history-of-quality/overview/overview.html (accessed June 17, 2011).
Bagian, J. P., C. Lee, J. Gosbee, J. DeRosier, E. Stalhandske, N. Eldridge, R. Williams, and M. Burkhardt. 2001. Developing and deploying a patient safety program in a large health care delivery system: You can’t fix what you don’t know about. Joint Commission Journal on Quality Improvement 27(10):522-532.
Chow-Chua, C., M. Goh, and T. B. Wan. 2003. Does ISO 9000 certification improve business performance. International Journal of Quality & Reliability Management 20(8):936-953.
Classen, D. C., and D. W. Bates. 2011. Finding the meaning in meaningful use. New England Journal of Medicine 365(9):855-858.
Classen, D. C., R. Resar, F. Griffin, F. Federico, T. Frankel, N. Kimmel, J. C. Whittington, A. Frankel, A. Seger, and B. C. James. 2011. “Global trigger tool” shows that adverse events in hospitals may be ten times greater than previously measured. Health Affairs 30(4):581-589.
Cohen, L. 1979. Innovation and atomic energy: Nuclear power regulation, 1966-present. Law and Contemporary Problems 43(1):67-97.
Cullen, D. J., D. W. Bates, S. D. Small, J. B. Cooper, A. R. Nemeskal, and L. L. Leape. 1995. The incident reporting system does not detect adverse drug events: A problem for quality improvement. Joint Commission Journal on Quality Improvement 21(10):541-548.
Dahiya, S., R. K. Khar, and A. Chhikara. 2009. Opportunities, challenges and benefits of using HACCP as a quality risk management tool in the pharmaceutical industry. Quality Assurance Journal 12(2):95-104.
DeRosier, J., E. Stalhandske, J. P. Bagian, and T. Nudell. 2002. Using health care failure mode and effect analysis: The VA National Center for Patient Safety’s prospective risk analysis system. Joint Commission Journal on Quality Improvement 28(5):248-267, 209.
FDA (Food and Drug Administration). 2009. The quality system regulation. http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/PostmarketRequirements/QualitySystemsRegulations/MedicalDeviceQualitySystemsManual/ucm122391.htm (accessed June 1, 2011).
Gardner, R. M., J. M. Overhage, E. B. Steen, B. S. Munger, J. H. Holmes, J. J. Williamson, D. E. Detmer, and the AMIA Board of Directors. 2009. Core content for the sub- specialty of clinical informatics. Journal of the American Medical Informatics Association 16(2):153-157.
Gastineau, D. A. 2004. Will regulation be the death of cell therapy in the United States? Bone Marrow Transplant 33(8):777-780.
Glasgow, J., J Scott-Caziewell, and P. Kaboli. 2010. Guiding inpatient quality improvement: A systematic review of lean and six sigma. Joint Commission Journal on Quality and Patient Safety 36(12):531-532.
Goodman, K. W., E. S. Berner, M. A. Dente, B. Kaplan, R. Koppel, D. Rucker, D. Z. Sands, P. Winkelstein, and the AMIA Board of Directors. 2011. Challenges in ethics, safety, best practices, and oversight regarding HIT vendors, their customers, and patients: A report of an AMIA special task force. Journal of the American Medical Informatics Association 18(1):77-81.
Grabowski, H. G., and J. M. Vernon. 1977. Consumer protection regulation in ethical drugs. American Economic Review 67(1):359-364.
Hauptman, O., and E. B. Roberts. 1987. FDA regulation of product risk and its impact upon young biomedical firms. Journal of Product Innovation Management 4(2):138-148.
HHS (Department of Health and Human Services). 2010. Health information technology: Initial set of standards, implementation, specifications, and certification criteria for electronic health record technology; final rule. Federal Register 75(144):44590-44654.
IOM (Institute of Medicine). 2011 (unpublished). Vendor responses—summary. Washington, DC: IOM.
ISO (International Organization for Standardization). 2011. Quality management principles. http://www.iso.org/iso/iso_catalogue/management_and_leadership_standards/quality_management/qmp.htm (accessed May 26, 2011).
Johansen, I., M. Bruun-Rasmussen, K. Bourquard, E. Poiseau, M. Zoric, C. Parisot, and M. Onken. 2011. A quality management system for interoperability testing. Paper presented at 23rd International Conference of the European Federation for Medical Informatics, Oslo, Norway, August 28, 2011, to August 31, 2011.
The Joint Commission. 2011. Sentinel events. Chicago: The Joint Commission.
Kim, D. U. 2002. The quest for quality blood banking program in the new millennium the American way. International Journal of Hematology 76(Suppl 2):258-262.
Laflamme, F. M., W. E. Pietraszek, and N. V. Rajadhyax. 2010. Reforming hospitals with IT investment. McKinsey Quarterly 20:27-33.
Leape, L. L. 2002. Reporting of adverse events. New England Journal of Medicine 347(20):1633-1638.
Marcus, A. A. 1988. Implementing induced innovations: A comparison of rule-bound and autonomous approaches. Academy of Management Journal 31(2):235-256.
NASA (National Aeronautics and Space Administration). 2011a. Confidentiality and incentives to report. http://asrs.arc.nasa.gov/overview/confidentiality.html (accessed April 18, 2011).
NASA. 2011b. Immunity policy. http://asrs.arc.nasa.gov/overview/immunity.html (accessed April 18, 2011).
National Academy for State Health Policy. 2010. Patient safety toolbox. http://www.nashp.org/pst-welcome (accessed June 24, 2011).
NIST (National Institute of Standards and Technology). 2006. Guide to integrating forensic techniques into incident response. Gaithersburg, MD: NIST.
NIST. 2010. Computerized common industry format template for electronic health record usability testing. Gaithersburg, MD: NIST.
NQF (National Quality Forum). 2011. NQF: Governance and leadership. http://qualityforum.org/About_NQF/Governance_and_Leadership.aspx (accessed June 24, 2011).
ONC (Office of the National Coordinator for Health Information Technology). 2011. Health IT workforce development program facts at a glance. http://healthit.hhs.gov/portal/server.pt?open=512&objID=1432&mode=2 (accessed June 24, 2011).
Quality and Safety Education for Nurses. 2011. Quality and safety competencies. http://www.qsen.org/competencies.php (accessed July 19, 2011).
Schneider, P. 1996. FDA & clinical software vendors: A line in the sand? Healthcare Informatics 13(6):100-102, 104, 106.
Shuren, J. 2010. Statement to IOM Committee on Patient Safety and Health Information Technology. Statement read at the Workshop of the IOM Committee on Patient Safety and Health Information Technology, Washington, DC.
Sittig, D. F., and D. C. Classen. 2010. Safe electronic health record use requires a comprehensive monitoring and evaluation framework. Journal of the American Medical Association 303(5):450-451.
Sittig, D. F., and H. Singh. 2009. Eight rights of safe electronic health record use. Journal of the American Medical Association 302(10):1111-1113.
U.S. Congress, Senate Subcommittee on Labor, Health and Human Services, and Education. 2000. Testimony of Leape, L.
VanRooyen, M. J., J. G. Grabowski, A. J. Ghidorzi, C. Dey, and G. R. Strange. 1999. The perceived effectiveness of total quality management as a tool for quality improvement in emergency medicine. Academic Emergency Medicine 6(8):811-816.
Veterans Affairs National Center for Patient Safety. 2011. FAQ: How do you define an intentional unsafe act? http://www.patientsafety.gov/FAQ.html (accessed August 9, 2011).
Walshe, K., and S. M. Shortell. 2004. Social regulation of healthcare organizations in the United States: Developing a framework for evaluation. Health Services Management Research 17(2):79-99.
Weeda, D. F., and N. F. O’Flaherty. 1998. Food and Drug Administration regulation of blood bank software: The new regulatory landscape for blood establishments and their vendors. Transfusion 38(1):86-89.
WHO (World Health Organization). 2005. WHO draft guidelines for adverse event reporting and learning systems: From information to action. Geneva: World Health Organization Press.












































