
Regulation, Reimbursement, and Public Health
Important Points Highlighted by the Individual Speakers
• The lack of a coherent oversight system could create a chasm between the use of genomic diagnostic tests and improved health.
• Collaboration among and within federal agencies could ease some of the limitations of the current regulatory system.
• Agencies have been experimenting with progressive approval as one way to provide more regulatory and reimbursement flexibility.
• A public health approach to genomic diagnostic tests would evaluate their utility to reach evidence-based recommendations and then evaluate their impacts at the population level.
Government has the responsibility to protect the public health and safety, yet it does so with a patchwork of laws and regulations and must base its decisions on evidence that is poorly developed in many areas. Three speakers discussed the approaches taken by the Centers for Disease Control and Prevention (CDC), FDA, and CMS. All acknowledged the many difficulties of overseeing genomic diagnostic tests while pointing toward promising innovations.
A 21ST-CENTURY OVERSIGHT SYSTEM
Major components of 21st-century medicine lack suitable oversight mechanisms, said Muin Khoury of CDC. Huge quantities of data have become available and much more is on the way, yet in many areas there remains an evidence gap between interventions and outcomes. Stakeholders have different perspectives on this evidence gap. While some may feel that sufficient evidence exists to meet their needs, others may not. The confusion generated by the lack of oversight creates less than optimal awareness and knowledge among consumers, providers, and systems.
In the area of genomic diagnostic tests, the lack of coherent oversight creates what Khoury termed “premature translation.” Genomic tests move from the bench to the bedside quickly with no strings attached because they go through the LDT route. “Spit in a test tube and you get results.” However, there remains a chasm between the use of these tests and improved health, which Khoury described as the “lost in translation” gap. Products seep through the translation process, some good and some bad, while information about their effectiveness is often lacking.
Khoury pointed to the need to develop what he called a public health approach to genetics (Figure 6-1). He admitted that the term is something of an oxymoron, since genetics is about personalized medicine and public
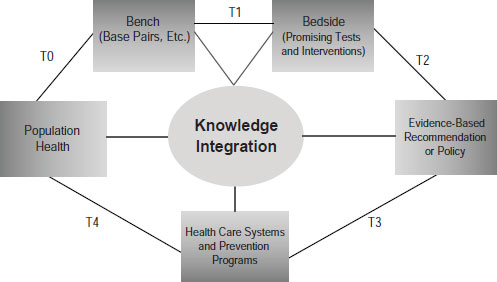
SOURCE: Khoury, IOM workshop presentation on November 15, 2011.
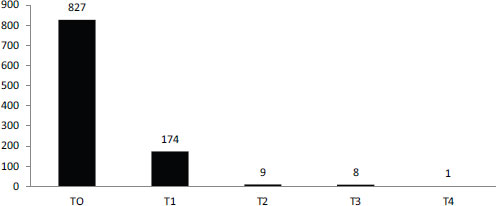
SOURCE: Schully et al., 2011.
health is about populations. But combining the two would benefit both endeavors through the development of a robust translational research enterprise that not only gets tests from bench to the bedside but evaluates their utility to reach evidence-based recommendations and then evaluates their impacts at the population level.
The current system does not fund this kind of research though, Khoury observed. According to a recent portfolio analysis of cancer genetics and genomic research at the National Cancer Institute, most funding goes for discovery or early translation (Figure 6-2). Less than 2 percent of funding is focused on clinical utility or later stages in the translation process (Schully et al., 2011). As a result, different stakeholders use different evidentiary frameworks to decide on the value of tests.
Potential Solutions
The way to solve this problem, according to Khoury, is through a 21st-century oversight system. For example, one such system would be the one suggested by Hayes, in which LDTs are eliminated and FDA oversees the approval of all tests. Another potential solution is greatly increased public and private funding for research focused on clinical utility and beyond. These may or may not be the right solutions “but at least [they are] outside the box.”
A third potential solution is a knowledge integration enterprise that would involve both information brokering and knowledge synthesis. One approach to knowledge integration has been pioneered by CDC’s Evalua-
tion of Genomic Applications in Practice and Prevention (EGAPP) group. EGAPP is an independent multidisciplinary working group that has been a “lightning rod” for discussion, according to Khoury, since it was formed in 2005. It has developed methods and outcomes processes, has conducted systematic reviews, has pointed to evidence gaps, and is beginning to tackle evidentiary standards for whole genome sequencing. “What EGAPP has tried to do is create analytical frameworks that allow data to be gathered across multiple platforms from observational studies to clinical trials,” Khoury said.
A second initiative launched by CDC and other public and private organizations in 2009 is the Genomic Applications Practice and Prevention Network (GAPPNet), which is designed to put stakeholders in the same room and connect them to data. The first objective of GAPPNet, according to Khoury, is to build the necessary information from discovery through health impacts. The second is to deal with stakeholder forces that affect translation by letting them talk through issues. “Maybe they will or will not reach consensus, but they need to be aided or helped by that oversight system.”
Overcoming Obstacles to Progress
The most important obstacle to the establishment of a 21st-century oversight system, said Khoury, is a lack of incentives. There are few incentives for public or private funding of research beyond the discovery phase, for knowledge synthesis and stakeholder convening, or for public and provider education.
To overcome these obstacles, it is necessary to start at the top. Pilot oversight “experiments” need to be developed and applied to deal with insufficient evidence, said Khoury. Public and private initiatives could come together to fund the generation of clinical utility evidence. The new Patient-Centered Outcomes Research Institute established by the Patient Protection and Affordable Care Act could potentially investigate these issues if it had sufficient funding. Small experiments by FDA, CMS, and the Agency for Healthcare Research and Quality (AHRQ) have moved in the right direction, but these need to be coordinated and expanded. As an example of a pilot project that Khoury initiated with funding from the American Recovery and Reinvestment Act of 2009, seven groups were funded to conduct comparative effectiveness research in genomics and personalized medicine and to develop a collaborative road map. Much of this work is about to be released. In addition, EGAPP continues to explore new methods and approaches. What is needed, said Khoury, is “a stakeholder-driven knowledge integration enterprise that explores novel methods of synthesis, decision analysis, and modeling.”
The 1976 Medical Device Amendments defined in vitro diagnostics as “medical devices” and established a risk-based regulatory paradigm for their oversight. The safety standard is that “there is reasonable assurance … that the probable benefits … outweigh any probable risks” [21 CFR 860.7(d)(1)]. The effectiveness standard is that “there is reasonable assurance that … the use of the device … will provide clinically significant results” [21 CFR 860.7(e)(1)].
The risk-based strategy has “strengths and weaknesses,” said Alberto Gutierrez of FDA. The standards are similar to those for drugs but they also differ in significant ways. For example, a premarket approval application (PMA) for a diagnostic is thought of by individuals to be similar to a New Drug Application in many ways, but controlled clinical trials are rarely submitted as the evidence base. Half of the devices that are currently on the market were not reviewed by FDA prior to their release and for another 40 percent, they just have to be similar to an already existing device. Only very high-risk devices require a PMA submission.
Gutierrez emphasized that the regulations extend from premarket to postmarket to compliance. In the premarket, industry provides the evidence and FDA reviews and clears or approves the device. In the postmarket, industry has the responsibility and FDA monitors and provides guidance. With compliance, FDA monitors companies to make sure that they comply with the law and regulations.
Progressive Approval
Gutierrez briefly discussed the idea of accepting a lower level of evidence premarket while relying on postmarket studies to gather additional evidence. FDA has done that in some cases, partly because the performance of a diagnostic is closely tied to the population in which it is used. Sometimes, good evidence for safety and effectiveness exists in one population but not another and, in this case, FDA will clear or approve the test but require postmarket data to be collected. For example, FDA cleared a test used in women with a pelvic mass that helps determine whether the mass should be removed by a gynecologist or by an oncologist,1 but it also required postmarket study to gather additional data on premenopausal women, for whom fewer data were available than for postmenopausal women.
![]()
1 Ovarian Adnexal Mass Assessment Score Test System; see http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm237299.htm.
Elements of Review
FDA evaluates analytic validity and clinical validity in its reviews, but Gutierrez stressed that the major factor that FDA considers is actually the intended use of a device, which does not necessarily preclude an evaluation of clinical utility. Some uses are very broad, in which case clinical utility is generally hard to assess, whereas others are very specific, in which case clinical utility is very important, said Gutierrez. If the claim behind an intended use is one of clinical utility, then that needs to be demonstrated. FDA also tries to be very transparent in its reviews, both consulting with expert panels when necessary and publishing the basis of its clearances.
Many groups, including FDA, recognize that there are regulatory gaps regarding LDTs, Gutierrez said, though they do not necessarily agree on how to solve these problems. Laboratories rightly observe that they are governed by CLIA. They also observe that clinical validity emerges from the published scientific literature, so that peer review is essential. However, “when the devices are very difficult to replicate, we’ve seen peer review and the literature not be a good form of regulation.”
A major problem with LDTs is that they have created confusion regarding the rules of the road from test development to research. How can patients be protected during postmarket research? How can it be determined that a test has failed, and what happens when a test has failed? “In general, this is an area we need to fix,” Gutierrez said.
Barriers to Successful Test Development
Gutierrez acknowledged the many problems raised by other presenters: diagnostic tests may not provide a sufficient return on investment; a lack of regulatory clarity can introduce uncertainty into the development of tests; many tests do not have much evidence regarding their utility; and LDTs lack standards and can be difficult to integrate into medical practice. These problems do not have easy solutions, said Gutierrez. Collaboration between FDA and CMS could help, and a pilot program between the two agencies is testing this approach. Collaboration within FDA also can be important. Much remains to be done in this area, but some of the collaborations within the agency are working well, according to Gutierrez.
FDA also has collaborated for many years with standards-setting bodies such as the National Institute for Standards and Technology. However, these efforts have been piecemeal and depend largely on finding someone who is willing to collaborate and the money to enable the collaboration. “It’s not an approach that is well thought out or that people can actually plan on in a very straightforward way.”
Putting the Patient First
The broader obstacles are that the health care system is “fairly chaotic,” with different people and institutions pulling in different ways. Financial interests favor the status quo, so change generally has to come from the political arena. However, this setting may not be optimal for discussions to identify and fix these issues, said Gutierrez.
Gutierrez concluded by pointing out that the focus should remain on the needs of patients. “We all need to figure out what is our responsibility in making this work.” People may need to take actions that are not in their best interest but are necessary to improve the overall system. “We all need to pull together, otherwise it’s not going to happen.”
The task of a test developer is to make investors, regulators, and users more confident about their test, said Louis Jacques of CMS. This task is made much easier when certain conditions apply.
First, it is easier when clear and consistent scientific evidence supports clinical utility, though this is a difficult condition to achieve, said Jacques. It also is easier when the risks of “medical misadventures” are known, measurable, and acknowledged. For instance, how easy is it for a physician to know that a genomic test result is mistaken or was not run on the proper sample? In addition, managing a perceived risk may affect an unknown or unrecognized risk. “If we arguably knew how to reduce our risk of heart disease by doing certain things, taking certain medications, how do we know that we haven’t increased our risk of neoplasm?”
Physicians need to consistently use the test where it fits in an overall management scheme, though this, too, is often difficult in practice. Even if CMS covered and paid for all genetic tests, they would probably be used chaotically in practice, Jacques said.
A standard nomenclature and taxonomy can increase confidence in the utility of a test. Having the relevant components consistently and precisely identified in a claims stream for a test would allow for easier evaluation. Currently, because of the use of stacking codes, Medicare already pays for many genetic tests, said Jacques, but “we’re not doing it in an intentioned or well-reasoned manner.” Rather, the test is part of a claims stream and is reimbursed unless someone prevents it.
Finally, a genomic test generates more confidence when there is agreement on its value. The evidence base is still largely immature, said Jacques. It stops well short of clinical utility, and at times short of analytic validity. Also, the evidence is not holistic, in that it is challenged to incorporate particular factors. For example, what is known about particular patient sub-
groups? “Is the relevance of a particular biomarker or a particular genetic test the same thing when you’re 70 years old and you’ve already expressed certain diseases as it is when you’re much younger?”
Factors in Coverage Decisions
Age is also a factor in assessing the value of genetic tests. Young people have a lifetime to manage their risks but may have little personal incentive to do so. Genomic testing may not be as relevant for a person who joins the Medicare program at 65 as it is when he or she is 2 years old, said Jacques. “Why shouldn’t people arrive … in the Medicare program with whatever predictive genetic factors that may be brought to bear, in fact, already done?”
Another factor Jacques cited is that genetic tests can have multiple platforms, multiple vendors, and multiple indications. In such a setting, reference standards can be critical. Several years ago, Jacques attended a meeting in which test developers could not agree on the definition of the colors used in their test. “I told them at that meeting that they had absolutely no chance of Medicare reimbursement unless they could at least agree on standards,” he said. “Sure enough, by the next year they had collaborated with NIST and actually developed standards.” Evaluating a product without knowing the starting reference point is a real difficulty, said Jacques.
Finally, a major challenge within CMS, as with FDA, is that payment decisions are binary. Statutes dictate how CMS must pay for covered health care practices. For example, congressional mandates delineate coverage for screening tests versus diagnostic tests, with screening tests tied to the findings of the USPSTF recommendations.
In contemplating the evaluation of tests, Jacques wondered if granting full reimbursement for a covered test would act as a disincentive to the development of further evidence. Jacques questioned whether it might make sense to pay initially at a lower level—say at 75 percent. Then, as the evidence base matured and if evidence demonstrated clinical benefit, a payment premium could be awarded—say 135 percent. Such a system could support future innovation and the development of “the next big thing.”
Innovation in Review
Several initiatives have been developed to enable collaboration between CMS and FDA, including the parallel review process and CMS representation on FDA’s Council for Medical Device Innovation. CMS has been open to accompanying test developers and others if they choose to meet with FDA for initial feedback, Jacques said.
CMS also has been doing coverage with evidence development for sev-
eral years, though it recently sought new public input on its CED guidance document. “I’m seeing big players in industry … make public comments that CED is good for innovation,” he said. Jacques also would like to see CED have greater breadth and flexibility so that not every new molecular indicator and LDT needs to be reviewed.
Medicare still has considerable local authority, Jacques pointed out, and local decisions do not necessarily apply nationwide. More collaborative review processes could help create greater nationwide consistency.
This page intentionally left blank.










