
In the second session, workshop participants considered the practical aspects of using genetics in the design and implementation of clinical trials, including the recruitment and stratification of trial participants and the effects on research costs and timelines. There have been a number of successes of precision medicine in the field of oncology, particularly with respect to previously intractable cancers such as melanoma and non-small cell lung cancer (NSCLC), said moderator Richard Schilsky, the chief medical officer at the American Society of Clinical Oncology. Increasingly, for a wide range of tumor types, it has been shown that understanding the genomic profile of the tumor and developing drugs targeted to oncogenic pathways can confer substantial benefits for those patients in whom the drug is appropriately used. However, a frequently encountered challenge in oncology is the development of drug resistance; in many cases targeted therapies do not result in long-term disease-free progression or other positive outcomes. Nevertheless, Schilsky said, the paradigm of precision medicine has been well proven in oncology as a way of delivering effective interventions for many patients, often with considerably less toxicity than chemotherapy.
Key principles and best practices for using genetics in clinical drug development were highlighted with four examples, two from the field of oncology and two from non-oncology areas. Roy Herbst, a professor of medical oncology and associate dean for translational research at Yale Cancer Center, described the Lung Master Protocol (Lung-MAP), a biomarker-driven study to identify new drugs with activity in a molecularly defined subsets of patients with advanced NSCLC. Zachary Hornby, chief operating officer at Ignyta, discussed the Studies of Tumor Alterations Responsive to Targeting Receptor Kinases (STARTRK-2) clinical trial, an innovative basket study of a drug to treat solid tumors with specific gene rearrangements. John Staropoli, associate medical director at Biogen, discussed genetic testing and clinical drug development for spinal muscular atrophy (SMA). Marni Falk, associate professor of pediatrics at the University of Pennsylvania, discussed using genetics in clinical trials for mitochondrial diseases.
LUNG-MAP: A BIOMARKER-DRIVEN UMBRELLA TRIAL FOR SQUAMOUS CELL LUNG CANCER
Lung cancer is the number one cause of cancer deaths worldwide (U.S. Cancer Statistics Working Group, 2016), Herbst said, noting that 20 years ago, the ceiling for cytotoxic chemotherapy had been reached and
new chemotherapies failed to significantly improve outcomes for patients (Schiller et al., 2002). It was clear, Herbst said, that new treatments were needed.
Searching for New Cancer Treatments for Lung Cancer
In 1998, ZD1839, an epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor later known in its generic form as gefitinib, was evaluated in a Phase 1 clinical trial in patients with metastatic non-small cell lung, head and neck, prostate, and colorectal cancer that was refractory to conventional chemotherapy or hormonal therapy (Herbst et al., 2002). About one in ten patients improved, which was an outcome unheard of at the time in metastatic disease, Herbst said. Although gefitinib was approved without a molecular correlate, it was discovered soon thereafter that specific mutations in the EGFR gene conferred sensitivity to this class of drugs, and the treatment landscape for lung cancer patients changed enormously. It allowed lung cancer to be thought of as a chronic disease, like heart disease or diabetes, Herbst said. As noted by Schilsky, a challenge for the treatment of cancer is the frequent development of drug resistance. In the case of resistance, Herbst said, the goal is to provide patients with a good quality of life as long as possible, while continuing to identify new molecularly targeted drugs to treat them as their disease becomes resistant.
A new approach was therefore needed for cancer clinical trials. In 2010, the Institute of Medicine (IOM) released the report A National Cancer Clinical Trials System for the 21st Century (IOM, 2010), which emphasized the critical need for a timely, large-scale, innovative clinical trials system. As summarized by Herbst, the report issued 12 recommendations to achieve four main goals:
- Improve the speed and efficiency of trial development and activation.
- Incorporate innovative science and trial design.
- Improve the prioritization, support, and completion of trials.
- Incentivize the participation of patients and physicians.
A number of concurrent efforts stemmed from the report, Herbst said, including the development of master protocols for lung cancer using innovative designs such as umbrella and basket designs, which prospectively employ enrichment strategies to incorporate molecularly defined subsets of patients (see Table 3-1). A master protocol can be used in umbrella or basket trial designs to allow experimental arms of the study to be added, dropped, or continued based on their performance (Herbst et al., 2002).
TABLE 3-1 Umbrella and Basket Trial Designs
| Trial Design | Approach | Examples |
|---|---|---|
| Umbrella | Test the impact of different drugs on different mutations in a single type of cancer |
|
| Basket | Test the effect of a drug or drugs on one or more mutations in a variety of cancer types |
|
SOURCE: Roy Herbst, National Academies of Sciences, Engineering, and Medicine workshop presentation, March 8, 2017.
Lessons Learned from the Lung-MAP Master Protocol Umbrella Trial: Viewpoint of a Clinical Trialist
After the discovery of lung tumor–associated EGFR mutations, an increasing number of oncogenic mutations were identified that further stratified lung cancer into molecularly defined subsets, Herbst said. The challenge, he explained, was to match those mutations with a targeted drug.
Researchers identified squamous cell carcinoma of the lung, which accounts for about 30 percent of NSCLC, as an area with a therapeutic unmet need and launched the Lung-MAP umbrella trial to address it. Lung-MAP was designed to
- develop drugs for uncommon/rare genotypes;
- use broad-based next-generation sequencing;
- achieve acceptable turnaround times for molecular testing for therapy (less than 2 weeks); and
- expedite the new drug-biomarker FDA approval process.
The ability of Lung-MAP to evolve as it progressed contributed to its overall success in quickly identifying and testing new targeted treatments, Herbst said. He offered a number of lessons learned from the trial, including
- choosing the right patient population;
- building an innovative public–private partnership;
- designing a feasible and adaptable protocol;
- enhancing mechanisms for accrual; and
- fostering broad participation.
Choosing the Right Patient Population
Squamous cell lung cancer was chosen as the NSCLC subpopulation for Lung-MAP for several reasons, Herbst said. First, screening for potential therapeutic targets is rapidly becoming a standard part of NSCLC treatment, and in 63 percent of squamous cell lung cancer cases, it is possible to identify potentially targetable mutations. Second, squamous cell lung cancer remains a subpopulation of NSCLC in which patient outcomes are poor, and there have been no substantial therapeutic developments in recent years.
Building an Innovative Public–Private Partnership
An important aspect of Lung-MAP, Herbst said, is that it was designed through a unique public–private partnership. Partners and collaborators include the National Cancer Institute (NCI), FDA, Friends of Cancer Research, the Foundation for the National Institutes of Health (FNIH), and SWOG (formerly the Southwest Oncology Group), all of which provide support for oversight and logistics. In addition, industry partners Pfizer, Genentech/Roche, AstraZeneca, MedImmmune, Amgen, Bristol–Myers Squibb, and Foundation Medicine provide funding and therapeutics for the trials as well as advisory contributions. Working with the NCI clinical trials cooperative groups allowed for maximum efficiency in enrolling patients with uncommon or rare, targetable mutations to facilitate analysis and FDA approval, Herbst said. Implementing a trial of Lung-MAP’s size and complexity took a village, he said. Lung-MAP has a unique organizational structure that enabled its success, including an oversight committee, an executive operations group, a project management office, and various committees and working groups to address such things as drug and biomarker selection, assay company selection, contracts, and data sharing.
Designing a Feasible and Adaptable Protocol
Lung-MAP is a multi-arm master protocol characterized by attributes that allow for feasible and adaptable protocol modifications and a rapid accrual of trial participants, Herbst said. The key features of the master protocol include
- homogenous, molecularly defined populations within each experimental arm, with consistency in eligibility criteria across arms;
- modular design where each arm is independent from the others;
- trial infrastructure that facilitates opening new arms quickly when an arm closes; and
- “seamless” trial design that allows a rapid transition from Phase 2 to Phase 3 for those sub-studies that meet their endpoints and an increased likelihood of detecting large treatment effects.
Though the trial has evolved over time to account for the removal or progression of trial arms that have failed or succeeded, respectively, or the addition of new arms as the treatment landscape evolves, it retains the same core structure (see Figure 3-1).
Patients are screened using the next-generation sequencing (NGS) platform for biomarkers of interest and, if positive for the biomarker, are enrolled in the appropriate biomarker-driven sub-study. Approximately 30 to 40 percent of patients are negative for all screened biomarkers and are therefore enrolled in a “non-match” sub-study to receive another investigational treatment, e.g., immunotherapy.
Over time, Lung-MAP has evolved to keep pace with a rapidly changing treatment landscape. For example, in 2015 (1 year after the launch of Lung-MAP), two immunotherapy agents were approved by FDA for the treatment of squamous cell lung cancer. Lung-MAP was adapted to take these agents
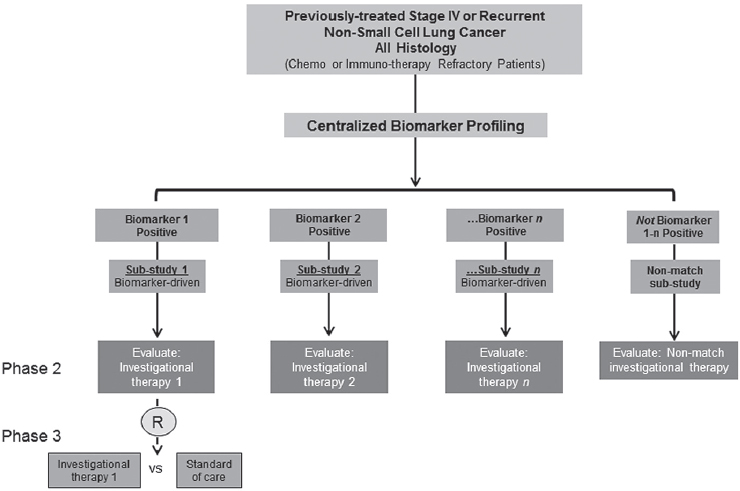
NOTE: R = registration trial.
SOURCE: Roy Herbst, National Academies of Sciences, Engineering, and Medicine workshop presentation, March 8, 2017.
SOURCE: Roy Herbst, National Academies of Sciences, Engineering, and Medicine workshop presentation, March 8, 2017.
into account, and immunotherapy has since become a major component of the non-match arms, Herbst said. Additional research is still needed, however, to identify which participants will respond to immunotherapies and if responses can be enhanced by combination therapies, he said. Finally, he noted, the trial design is routinely reviewed and modified based on input from academic researchers, physicians, basic scientists, and, most importantly, patients.
Enhancing Mechanisms for Accrual
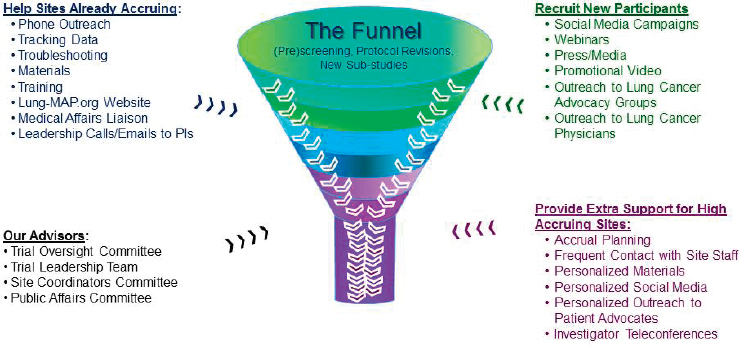
When researchers are seeking to identify genomically driven populations, they must screen many patients to identify the few who present with the biomarker of interest. To address this, the Lung-MAP leadership created an accrual enhancement committee to help existing sites recruit additional participants and to provide additional support to high-accruing sites through innovative outreach, such as the use of social media, webinars, or promotional videos (see Figure 3-2). Herbst pointed out that the accrual enhancement committee, in order to maximize overall accrual, chose to prioritize already-accruing sites rather than focusing time and resources on sites that were not accruing. He shared examples of the informational fliers provided to patient advocacy groups and showed a brief promotional video, all of which can be found on the Lung-MAP website.1 Given that patients
___________________
1 Infographics are available at http://www.lung-map.org/media/press/lung-map-infographics-illustrate-how-trial-helping-patients. The video shown can be viewed at http://www.lung-map.org/about-lung-map (accessed April 26, 2017).
with metastatic lung cancer have a high probability of failing first-line treatments, the Lung-MAP leadership recognized that screening patients for trial enrollment at disease progression was not feasible and therefore added an additional pre-screen option to the trial protocol, Herbst said. He explained that patients can be pre-screened while still on first-line chemotherapy and can pre-enroll for the trial so that they are ready to participate should their cancer progress.
Fostering Broad Participation
There are now more than 740 sites participating in the Lung-MAP trial, including several in Canada, Herbst said. At the time of the workshop, more than 1,100 patients were registered, with more than one-third of those having been prescreened and over 800 assigned to one of the sub-studies.
Adequate funding is needed to secure site participation for this number of participants, Herbst said, and additional funds and reimbursements were given to sites, as needed, for conducting the trial and supporting research staff. Furthermore, because most cancer patients are treated in their community, not at major academic centers, the trial needed to be community based. This allows researchers to reach underserved areas, engage patients and physicians, and bring the therapies under study to the patients, Herbst said.
STARTRK-2: A BASKET TRIAL OF ENTRECTINIB FOR THE TREATMENT OF SOLID TUMORS WITH SPECIFIC GENE REARRANGEMENTS
As discussed by Herbst, there are two key frameworks for biomarker-enabled clinical trials: umbrella and basket, with Lung-MAP representing the former. An example of the latter, a basket study termed STARTRK-2,2 was discussed by Hornby. STARTRK-2 is evaluating a single drug (entrectinib) for efficacy against multiple solid tumor histologies and lymphomas expressing a small subset of biomarkers.
Entrectinib is a selective inhibitor of five protein targets: c-ros oncogene 1 (ROS1), anaplastic lymphoma kinase (ALK), and three neurotrophic tyrosine receptor kinases (TRKA, TRKB, TRKC). These five proteins are cell-surface receptors prone to genetic translocations that lead to
___________________
2 For more information Studies of Tumor Alterations Responsive to Targeting Receptor Kinases (STARTRK-2), An Open-Label, Multicenter, Global Phase 2 Basket Study of Entrectinib for the Treatment of Patients with Locally Advanced or Metastatic Solid Tumors that Harbor NTRK1/2/3, ROS1, or ALK Gene Rearrangements (NCT02568267), see https://clinicaltrials.gov/ct2/show/NCT02568267 (accessed April 26, 2017).
fusion proteins, Hornby explained. The fusion proteins constitutively activate downstream oncogenic signaling pathways, such as MAP kinase, PI3 kinase, and PLC gamma. Entrectinib is the most potent TRK and ROS1 inhibitor in clinical development, he said. Entrectinib binds to and inhibits the fusion proteins and downregulates downstream oncogenic signaling, resulting in rapid tumor response in preclinical models and select patient populations. Importantly, he said, entrectinib was also designed to cross the blood brain barrier, a key consideration in the development of oncology drugs in order to address primary brain malignancies and secondary central nervous system (CNS) metastases. Through a broad survey of available tumor genomic data, TRK fusion proteins were identified in more than 30 different tumor types; however, within any given tumor histology, these gene fusions are generally quite rare.3 Notably, entrectinib exhibits high anti-proliferative activity regardless of the fusion partners and, importantly, regardless of the tissue of origin, Hornby said, citing results from two recent Phase 1 studies that showed a 79 percent objective response rate in patients harboring tumors with NTRK, ROS1, or ALK gene fusions (Drilon et al., 2017). The response rate was consistent across each of the molecular targets, TRK, ROS1, and ALK. In other words, he said, entrectinib is active against multiple cancer types, as long as the relevant mechanism is present.
Multifactorial Design of the STARTRK-2 Basket Trial
A challenge for precision drug development, Hornby said, is how to design a registration-enabling clinical program and accompanying IVD approach that can identify and enroll genetically identified patients who are individually rare within a given tumor histology but collectively numerous. Taken separately, no individual tumor histology would have sufficient patient numbers to complete accrual in a molecular subtype for a “traditional” clinical trial. Adding to this challenge, patients presenting with many of the relevant tumor types for entrectinib (e.g., head and neck cancers) are not frequently genetically tested at the time of diagnosis, and when genetic testing does occur, biomarkers of interest for entrectinib (e.g., NTRK) may not be on the standard panels. A solution to these issues was built into the design of STARTRK-2, which employs a flexible, multi-pronged diagnostic workflow and trial design with the following characteristics.
___________________
3 There are some exceptions where these gene fusions can be quite common in very rare tumors, for example in mamma-ry analog secretory carcinoma, or secretary breast cancer, where incidence of fusions can be over 90 percent.
Biomarker-Driven Recruitment
The STARTRK-2 trial is open to patients with all solid tumor histologies and lymphomas that harbor the requisite biomarker (NTRK, ROS1, or ALK gene fusions). Genetic screening can occur at any point in a patient’s care, Hornby said, even when the patient is in active treatment and responding to another drug.
Innovative Mutation Detection Approach
Ignyta has taken a three-pronged approach to enable broad diagnostic testing to identify relatively rare patients for enrollment by
- establishing a central laboratory that is Clinical Laboratory Improvement Amendments (CLIA) certified and College of American Pathologists (CAP) accredited and that performs highly sensitive fusion testing through Ignyta’s proprietary Trailblaze Pharos™ assay;
- working with diagnostic consortia and regional commercial laboratories throughout the United States, Europe, and Asia, including pan-national initiatives like Screening Patients for Efficient Clinical Trial Access in Advanced Colorectal Cancer (SPECTAcolor4), to ensure that testing of relevant biomarkers for the STARTRK-2 trial are added to diagnostic panels and that patients benefit from an established infrastructure; and
- working to enable academic medical centers throughout the United States, Europe, and Asia to perform high-sensitivity fusion testing.
In developing an IVD, Hornby emphasized, it is important to interact with regulatory agencies very early on in the drug development process. Although the standard pathway for targeted therapies is that the new drug application (NDA) is submitted in parallel with a premarket approval (PMA) application for the diagnostic assay, he said that it was critical to the trial design that, from initiation, there was a reliable central assay that would be used to recruit and enroll the proper patient population.
Liberal Eligibility Criteria
Overall, the inclusion and exclusion criteria for the trial are restricted only to what is necessary to inform safety and efficacy testing, Hornby said. Specifically, the inclusion and exclusion criteria include a broad range
___________________
4 For more information about SPECTAcolor, see http://spectacolor.eortc.org/about (accessed May 15, 2017).
of tumor histologies, prior treatment histories, and age ranges and do not include traditionally restrictive criteria related to renal or hematological function or primary or secondary CNS disease. The primary analysis is restricted to a defined patient population, however, in order to fulfill the analytical requirements needed to assess whether the trial reaches its primary endpoint. As such, Hornby said, certain patients may contribute only to a subset of endpoints, such as progression-free survival, overall survival, safety, or pharmacokinetics.
Global Catchment and Just-in-Time Trial Sites
Two key aspects that aid in STARTRK-2 accrual are the global reach of the catchment area and the “just-in-time” network, which can be activated when a new patient is identified in the area, Hornby said. At the time of the workshop, there were more than 150 clinical sites across 14 countries on four continents, including more than 70 established clinical sites in the United States and an additional 150 clinical trial sites in the United States that can be activated “just in time” when a new patient is detected in that area. This is facilitated via a site management organization using a central institutional review board (IRB) and central contracting process to expedite enrollment. Additionally, a travel concierge is made available to assist patients who are not near a clinical site to travel to the nearest site for treatment. To demonstrate the power of the collaborative just-in-time network and the role of the commercial diagnostic screening partner and site management organization, Hornby presented a case study of a patient who was enrolled in the STARTRK-2 trial within 7 days of diagnosis of an actionable mutation (ROS1+) (see Box 3-1).
Pooled Statistical Analysis
A critically important component of the trial design for analyzing trial data, Hornby said, is that each “basket” represents the combination of a target gene fusion and a tumor histology or lymphoma (e.g., ROS1+ in NSCLC). All baskets can contribute to the primary endpoint (objective response rate) through pooled statistical analysis. This approach could lead to a “molecular label indication,” Hornby said, where the indication is not defined by the tumor histology, but rather by the presence of a specific genetic aberration.
Parallel Drug and Diagnostic Development
Developing targeted oncology drugs and IVDs in tandem takes an integrated approach to therapeutic and diagnostic development, Hornby
said. An internal diagnostics capability allows for the illumination of the molecular drivers of cancer, for quickly advancing the most appropriate molecularly targeted therapies to address these drivers, and for more seamlessly translating these discoveries to the clinic, he said. For example, the Trailblaze Pharos™ assay was developed in collaboration with FDA’s Center for Devices and Radiological Health and has been granted an investigational device exemption and an expedited access pathway designation by FDA. The IVD will also be the basis for a PMA (for Class III medical devices) to be submitted in parallel with an NDA, if applicable, for entrectinib.
Patient and Provider Awareness
Ignyta is working with disease and patient advocacy groups to raise awareness of the STARTRK-2 study and of the potential importance of genetic testing. Outreach strategies include presenting and sharing live data updates at conferences; issuing newsletters, banner ads, and blast emails; conducting search engine optimization to provide informed patients with genetic diagnoses an avenue for finding the study; and maintaining an up-to-date clinical trial website tailored to both providers and patients.
Compassionate Use
As an example of the trial sponsor’s willingness to go outside of the trial design to provide expanded access (compassionate use) to at-need populations, Hornby described the use of entrectinib in a 20-month-old patient with a rare, congenital, recurrent, metastatic infantile fibrosarcoma harboring an NTRK3 gene rearrangement that might be responsive to entrectinib. The patient presented with metastasis to the lungs, and after 1 year of chemotherapy the disease returned in the brain and salvage chemotherapy was ineffective, Hornby said. A single-patient expanded access protocol was designed by working with FDA. At that time, a pediatric formulation of entrectinib was not yet available, so capsules were broken open and mixed into baby foods to deliver the drug. At baseline, the patient had a major cystic lesion in the brain, significant edema, and was minimally responsive and not eating. At day 35 on entrectinib, there was a reduction in the lesion size, reduction in swelling, normalization of the brain midline, and importantly, the patient was eating, mobile, and more alert, Hornby said.
Lessons Learned: Viewpoint from Industry
In closing, Hornby summarized several lessons learned from the STARTRK-2 clinical trial thus far:
- Pursue rare patient populations that are highly actionable.
- Design clinical trials to match the underlying disease biology and the mechanism that the drug targets.
- Engage regulatory agencies early, and work collaboratively, if possible.
- Consider building, rather than buying, when a ready-made solution does not exist, but adapt as the landscape evolves.
CLINICAL DRUG DEVELOPMENT FOR SPINAL MUSCULAR ATROPHY
SMA is the leading monogenic cause of mortality in infants and toddlers, with an incidence of about 1 in 10,000 live births, Staropoli said. Its pathology is uniformly caused by a loss of function of the survival motor neuron 1 gene (SMN1), which results in progressive degeneration of the alpha motor neurons of the spinal cord. Disease manifestations include progressive muscle weakness and atrophy, gastrointestinal disturbances and swallowing dysfunction, and respiratory infections from late-stage failure of the major muscles of respiration. Despite having a common underlying genetic defect, SMA has a broad phenotypic spectrum, driven largely
by the copy number of SMN2, which encodes mostly transcripts lacking exon 7, but naturally produces a small percentage (10−20 percent) of correctly spliced transcripts that serve to functionally compensate for the nonfunctional SMN1 gene. The first FDA-approved drug for SMA, nusinersen (Spinraza), was made available in December 2016. Nusinersen is an antisense oligonucleotide-based splicing modifier that promotes proper splicing of the backup SMN2 gene, thereby increasing the amount of functional transcript and alleviating some of the symptoms of the disease. Knowledge of the underlying biology of SMA was critical for the development of nusinersen as a treatment, Staropoli said.
Developing a Treatment and Diagnostic for SMA: A Case Study of Nusinersen
Using the underlying biological knowledge of SMA, the drug nusinersen was designed to target the splice silencer element known as ISS-N1. Silencing this genomic sequence promotes exon 7 inclusion in SMN2 transcripts so that >50 percent (compared to 10−20 percent) of SMN2 transcripts are full-length. In pre-clinical studies, the timing of the treatment with nusinersen was shown to be critical for therapeutic effect. In an induced mouse model of SMA, earlier treatment lessened the disease phenotype and rescued about two-thirds of gene expression changes, while delayed introduction of nusinersen resulted in successively less rescue of the gene expression profile (Staropoli et al., 2015). Subsequent clinical trials showed results that were comparable to the preclinical animal studies—specifically that presymptomatic intervention yielded better outcomes than post-symptomatic intervention. A subsequent open-label trial in which presymptomatic neonatal subjects (less than 6 weeks of age) were treated with nusinersen resulted in a robust increase in neurologic function compared with early-onset (>7 months) treatment or placebo (sham) control, Staropoli said. These results emphasize the importance of identifying and treating patients as early as possible, he said, adding that he is optimistic about the potential to shift the treatment paradigm for SMA from symptomatic treatment to early, pre-symptomatic treatment. In aggregate, the preclinical and clinical data to date support the case for newborn genetic screening, he added.
Next Steps for SMA: Newborn Screening
Biogen, in collaboration with the Centers for Disease Control and Prevention and colleagues at National Taiwan University in Taipei, developed and validated an assay that distinguishes between SMN1 and SMN2 and that can detect the absence of SMN1 (Taylor et al., 2015). The assay uses dried blood spots and can be multiplexed with the DNA-based newborn
screening test for severe combined immunodeficiency. Pilot studies of newborn screening are under way, Staropoli said. As of June 2016, more than 100,000 newborns had been screened in Taiwan, 6 of whom were found to have SMA and 2 of whom chose to enter clinical trials; 2,000 newborns had been screened in New York State, one of whom was found to have SMA. Thus far, there have been no verified false negatives, he added. Recently, an application was submitted to the U.S. Department of Health and Human Services for inclusion of the SMA assay on the U.S. Recommended Uniform Screening Panel, supported by preclinical and clinical data showing that earlier treatment yields better outcomes.
USING GENETICS IN CLINICAL TRIALS FOR MITOCHONDRIAL DISEASES
Mitochondrial diseases are highly heterogeneous genetic diseases, whose pathogenesis results from a dysfunction in respiratory chain activity and corresponding reduced cellular energy production. Despite the inherent complexity of both the genetics and clinical presentation of mitochondrial disease, the field has been propelled forward over the past 25 years by advances in the basic and translational sciences, Falk said. There is also an activated and empowered patient community and a high-functioning and influential disease advocacy organization, she said. Collaboration within the mitochondrial disease community to increase education, promote funding for research, provide patient support, and conduct clinical research with the end goal of finding treatments or cures has also positively affected the field.
Identifying Causative Mutations for Mitochondrial Disease: The Power of Next-Generation Sequencing
There are many genetic causes of mitochondrial disease across both the mitochondrial and nuclear genomes, Falk said, and the molecular understanding of mitochondrial disease is rapidly evolving. For example, a decade ago, mutations in only about 70 different genes were known to cause mitochondrial disease, while there are now at least 300 different known causative genes across the dual genomes (Koopman et al., 2012). Collectively, mitochondrial diseases are the single most common group of inborn errors of metabolism, affecting at least 1 in 4,300 people of all ethnicities and ages.
Because of the genetic complexity inherent to mitochondrial disease, there is no common, pan-disease biomarker. However, the last decade has seen a growing understanding of the various molecular pathways that can contribute to the pathogenesis of mitochondrial disease. The current scientific characterization of mitochondrial disease has evolved from an organ-
based disease characterized by multi-system organ failure, to an energetic disease resulting from dysfunction of oxidative phosphorylation, to the present understanding of it as a pathway-based disease with some known molecular etiologies. Falk suggested that this deeper understanding could provide potential molecular targets for intervention and that these different, individually rare diseases could potentially be grouped into cohorts that might each respond to a specific therapy.
NGS has revolutionized both the discovery of novel pathogenic genetic mutations and the confirmation of clinical diagnosis, Falk said, and she highlighted two resources specifically developed for drug discovery and development efforts. First, the GENESIS software tool allows researchers to upload genomic data from individuals with mitochondrial disease and compare them to genomic data in the general population.5 Another resource, the Mitochondrial Disease Sequence Data Resource,6 provides a common place for publicly curated data about genes that cause mitochondrial disease and access to tools for data mining and analysis (Falk et al., 2015). These resources are important for collating genomic data in a central location, where they can serve as an invaluable tool for elucidating the underlying genetic causes of mitochondrial disease, identifying modifiers of disease severity, guiding drug discovery efforts, and informing clinical drug development programs, Falk said.
During an ensuing panel discussion, Schilsky observed that the definitive diagnostic test for mitochondrial disease—whole genome sequencing—is costly and not widely available at the point-of-care for most health care providers. He added that deciding when a patient needs to be screened for a mitochondrial disorder can be challenging. There is a useful resource for primary care providers that provides guidance for recognizing and diagnosing mitochondrial disease (Haas et al., 2007), Falk said, which is important because there is a shortage of clinical geneticists and metabolic doctors in the United States who are able to recognize these patients. Typically, she said, mitochondrial disease is progressive and involves more than one organ system, so primary care providers should be educated to look for multiple system dysfunction and refer patients to testing for mitochondrial disease if indicated by the patient’s clinical presentation. Whole-exome sequencing or targeted NGS analyses of nuclear and mitochondrial gene candidates are important and necessary tools in rare disease diagnosis, Falk said. People with undiagnosed disorders should be afforded more access to this type of
___________________
5 For more information on the GENESIS software tool see http://thegenesisprojectfoundation.org (accessed May 15, 2017)
6 For more information on the Mitochondrial Disease Sequence Data Resource (MSeqDR) see https://mseqdr.org (accessed May 15, 2017).
technology, she added, but it should be used judiciously and in conjunction with the clinical history.
Developing a Community of Public and Private Partners to Find a Treatment for Mitochondrial Disease
There are no proven, effective therapies for mitochondrial disease, Falk said, noting that it is individually very rare and highly heterogeneous with respect to both causative genetic mutations and clinical phenotypes. However, she said, over the past 25 years there has been a series of rapid advances in basic and translational science and diagnostic techniques, while at the same time patients, who are essential participants in the research process, have taken on a more centralized role. The genesis of a patient-driven movement began in the early 1990s when mitochondrial disease advocacy groups coalesced under the umbrella of the United Mitochondrial Disease Foundation (UMDF). The UMDF’s initial mission was to provide an understanding and spread awareness of mitochondrial diseases, but the mission now includes providing critically important funding and resources to support research, such as the previously described Mitochondrial Disease Sequence Data Resource.
In addition to these patient-driven efforts, many other partners have shaped the mitochondrial disease community, Falk said. The North American Mitochondrial Disease Consortium, a member of NIH’s Rare Disease Clinical Research Network, maintains a patient registry for natural history studies and has established a biorepository to facilitate the sharing of biospecimens, including DNA and cell lines, among consortium members. Biopharmaceutical companies are engaged in drug development and clinical trials, and there are numerous medical centers, clinicians, and researchers also engaged in drug discovery and clinical research efforts, she said. Government research agencies, including NIH and the U.S. Department of Defense, are involved in mitochondrial basic science and disease research, and lobbyists and legislators advocate for increased research funding support (Camp et al., 2016). FDA provides critical support and guidance related to regulatory science and in October 2015 participated in a Critical Path Innovation Meeting,7 said Falk, alongside representatives from the biopharmaceutical industry and academic researchers, to provide guidance on how to best approach developing drugs for patients with mitochondrial disease.
___________________
7 For more information about the Critical Path Innovation Meeting see the summary at https://ods.od.nih.gov/attachments/CriticalPathInnovationMeetingSummary.pdf (accessed June 5, 2017).
Mitochondrial Disease Clinical Trials
Few clinical trials have been conducted to date for mitochondrial disease, Falk said, although some trials have recently been launched that focus on common clinical outcomes in genetically confirmed mitochondrial disease cohorts. In emphasizing the willingness of mitochondrial disease patients to participate in clinical research, Falk described results from a survey that measured both patients’ symptom frequency and their corresponding interest in participating in clinical trials.8 One detail from the survey that would likely be of interest to those designing patient-centered mitochondrial disease clinical trials is that patients are most willing to participate in clinical research for their top five most common symptoms: muscle weakness, chronic fatigue, exercise intolerance, gastrointestinal problems, and balance problems, said Falk.
Novel Clinical Trial Models for Bringing Effective Therapies to Patients with Mitochondrial Disease
Historically, the translational pathway for moving from basic science discoveries to clinically relevant treatments has involved defining the disease, prioritizing treatment outcomes, identifying potential treatment options, designing a trial with a treatment(s) that addresses those outcomes, and, if the trial is successful, moving the approved therapeutic into the clinic as a standard of care (see Figure 3-3).
However, this classical one-size-fits-all approach is potentially not the most conducive for bringing precision treatments to market for rare diseases, including mitochondrial disease, Falk said. An important step in the drug development pathway for mitochondrial diseases is in vitro drug testing, which includes studies conducted with primary patient cells and genetic models, including integrated physiologic endpoints and toxicity studies within the altered metabolic environment of each disease subtype. Since dysfunction in the mitochondria can have wide-ranging and disparate effects on cellular function, depending on the causative mutation(s), drugs may behave differently than they would in non-diseased cells or across different disease subtypes, she said. A better understanding of the molecular aberrations underpinning mitochondrial disease and the subsequent mechanistic defects at play could also facilitate preclinical in vitro laboratory drug testing and inform the design of targeted clinical trials in molecularly defined participants.
___________________
8 The survey that Falk described at the workshop is currently unpublished, but is in preparation for publication (Zolkipli-Cunningham et al.).
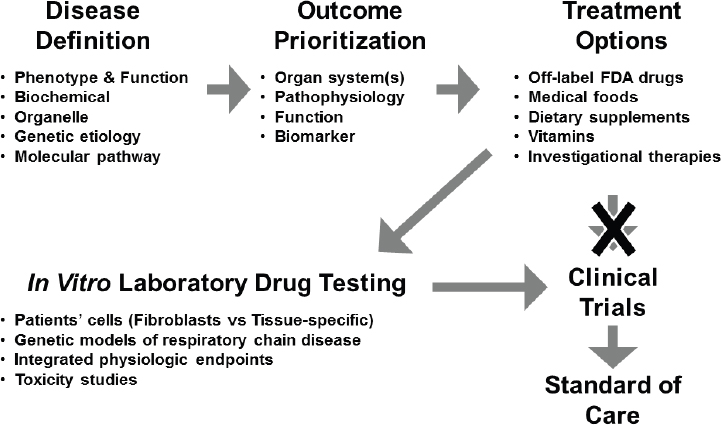
NOTE: FDA = U.S. Food and Drug Administration.
SOURCE: Marni Falk, National Academies of Sciences, Engineering, and Medicine workshop presentation, March 8, 2017.
Enrolling sufficient numbers of participants in trials for rare diseases can be challenging, Falk said, pointing out that FDA has produced guidance on this issue.9 To address challenges presented by conducting traditional clinical research in rare diseases, Falk described the emerging concept of performing separate single-patient, or “N-of-1,” clinical trials. N-of-1 trials could be designed to test therapies that were experimentally validated in vitro and found to be most effective in the corresponding mitochondrial disease patient cell line or animal model. Conducting multiple blinded, cross-over studies in single patients is a viable clinical trial approach to consider for mitochondrial diseases, Falk said, because patients typically vary markedly from one another, patients’ medical conditions vary over time, the optimal treatment(s) may differ between patients, and there are too few similar patients to pool for clinical research.
Finally, Falk said, it is important to conduct trials for mitochondrial diseases in both adults and children. At least one-third of mitochondrial disease
___________________
9 To view the FDA draft guidance, Rare Diseases: Common Issues in Drug Development, see https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM458485.pdf (accessed May 7, 2017).
patients are children, she said, and disease phenotypes can vary widely, even in the same mutation type and between children and adults.
Needs and Opportunities in Rare Disease Precision Medicine
Most rare diseases have no effective treatments or cure, Falk said, adding that there is a pressing need to clarify the causes and the consequences of individual diseases. It will be important to test the effects of treatments in relevant cellular and animal models of genetic diseases before initiating trials in patients. Furthermore, she urged rare disease researchers to consider how the mechanism of a rare disease might overlap with those of chronic or complex disease as a way to garner broader interest for research funding.
The opportunity exists to improve outcomes for rare disease patients by using precision medicine to target underlying molecular aberrations, Falk said, and she provided her perspective on what is needed to make advances in this area:
- Train clinicians to diagnose, care for, and effectively manage complex patients.
- Validate common biomarkers to diagnose and monitor disease in subgroups.
- Perform natural history studies to understand the spectrum of the disease.
- Unite experts to maximize meaningful studies of existing and emerging therapies.
- Partner with patient advocacy groups to develop treatments for prioritized problems.
- Integrate pharmaceutical, academia, and government resources to lower barriers and channel limited resources into meaningful studies that address disease mechanisms and develop efficacious treatments for distinct disease sub-groups.
- Consider innovative N-of-1 precision trials tailored to each rare disease patient or subgroup.




















