
3
Factors Influencing Affordability
The affordability of prescription drugs in the United States is influenced by a complex and highly interactive set of factors. The factors that tend to increase the cost of drugs for patients include the following, each of which is discussed in turn in this chapter:
- High launch prices, with the price of the drug then often increasing over time.
- Inadequate competition when market exclusivity ends.
- The interaction of market power, health insurance, and the lack of effective incentives for controlling product price.
- Unequal bargaining power between buyers and sellers.
- Research, development, and marketing expenditures as well as other business expenses.
- Insurance benefit designs with significant patient cost-sharing provisions.
- Inadequate performance of patient assistance programs and other public programs intended to make medicines more affordable for patients.
- Lack of adequate information affecting choices regarding medicines.
PRODUCT PRICING
Patent law establishes the exclusive right for inventors to apply their work for a specified period of time, either through direct manufacturing or by licensing to others. During patent exclusivity, prices of products are typically
set higher that permit patent holders to realize greater profits than would be achievable in a competitive marketplace. Many observers of the biopharmaceutical sector characterize this pricing practice as “what the market will bear.” Others rationalize it as properly and necessarily rewarding the pursuit of a high-risk, capital-intensive endeavor. That is, producers can be expected to set prices that are constrained only by how much consumers are willing to pay for a product that is protected by exclusivity.
The entry of generic drugs into the market provides competing products that are generally sold at much lower prices than the original branded product. Although the original branded product may continue to be offered at a high price, typically the lower cost of the generic will lead a large number of consumers to choose it. In recent years, between 80 and 90 percent of all prescriptions in the United States have been filled with generic products (Boehm et al., 2013; GPhA, 2015; Lee et al., 2016). However, if there is insufficient competition among the generic alternatives themselves, the prices of a drug might not drop to the anticipated competitive level. This particular issue is explored in a later section of this report.
A parallel issue relates to the particular manner in which drugs are actually priced in the United States (as described in Chapter 2). Specifically, manufacturers and distributors of drugs start with list prices at the time of launch and often modify them over time. However, in many cases a product’s list price is immediately discounted by manufacturers and distributors in sales to pharmacy benefit managers (PBMs), insurance companies, wholesalers, retailers, and others. Unfortunately, there are few publicly available data about the nature of these discounts, so it is impossible to determine exactly how net prices for consumers are derived. The situation is made more complex by the fact that the net price may differ greatly among different consumers.
Biopharmaceutical manufacturers often state that too much attention is focused on the list price of drug, as opposed to the end cost to health plans and patients. However, in a system with a broad use of rebates and discounts, list price matters because it is the starting point for all negotiations in the supply chain. As discussed earlier, it also matters because (1) uninsured patients pay list price at the pharmacy, and (2) cost sharing for insured patients is sometimes defined as a fraction of list price. The effects of high list prices are discussed in the following sections, with branded, generic, and other drug products covered separately.
Branded Drugs
Although branded medications make up approximately 10 percent of all prescriptions in the United States, they account for nearly three-quarters
of prescription drug spending (GPhA, 2015). Spending for all retail prescription drugs accelerated significantly in 2014 and 2015, before slowing in 2016 (QuintilesIMS, 2017a). The spending rate was 10.3 percent, which rose to 12.4 percent between 2014 and 2015 before falling to 5.8 percent in 2016—still twice the 2.5 percent rate of growth in 2013 (QuintilesIMS, 2015a, 2016a, 2017a).
The cost of branded drugs is influenced by their launch prices—the prices set by the manufacturer for the new drugs when they first become available on the market—and the subsequent annual increases in their list prices. Recent data on anti-cancer drugs show that on average launch prices increased by about $8,500 per year over the past 15 years (Howard et al., 2015). Other studies have found similar increases in the prices of cancer drugs after their launch (Bach, 2009; Bennette et al., 2016; Shih et al., 2017).
A 2009 report from the U.S. Government Accountability Office (GAO) estimated that between 2000 and 2008, 416 brand-name drug products displayed “extraordinary” price increases (GAO, 2009). The 416 products represented 321 specific medications, with some medications being available in different drug strength and dosage forms; for example, the 416 products included eight different strength and dosage forms of the beta blocker Inderal. Most often the increases in price reported in the study were between 100 and 499 percent, but in a few cases, specifically for drugs used to treat such conditions as fungal or viral infections or heart disease, a drug’s price increased by 1,000 percent or more.
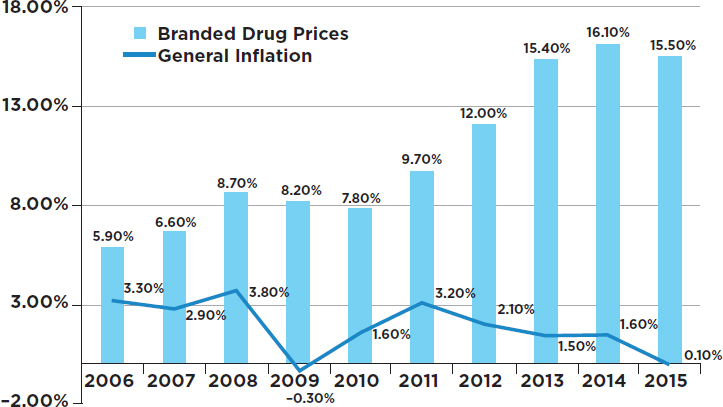
The absolute price increases for branded drugs ranged from $0.01 per unit to $5,400 per unit. The unit price of a drug is, of course, only one factor in determining the cost of a full course of treatment for a medical condition. The cost for a full course of treatment for one drug used to treat one rare form of cancer increased from $390 to more than $3,000 during the study period (GAO, 2009). Figure 3-1 shows how the prices of 268 top branded drugs rose throughout the period 2006–2015, with the yearly increases being consistently higher than the increases in the overall consumer price index—sometimes much higher.
Spending on specialty medicines has nearly doubled over the past 5 years, clearly outpacing the consumer price index and accounting for more than two-thirds of the overall growth in spending on medicines between 2010 and 2015 (AHIP, 2015; QuintilesIMS, 2016a). One result of this increase is that Medicare beneficiaries face rapidly growing out-of-pocket payments for specialty drugs. This trend is likely to continue as the population ages and more treatments become available for difficult-to-manage diseases (Dusetzina and Keating, 2015; Dusetzina et al., 2017; Trish et al., 2016). On the challenge of how to go about financing very expensive
SOURCE: Schondelmeyer and Purvis, 2016.
branded drugs, see Box 3-1. Whether existing or new drug therapies are actually effective in patients is another issue that must be considered.1
Generic Drugs
Once branded medications lose their patent exclusivity, generic versions can enter the market with approval from the U.S. Food and Drug Administration (FDA). Generic drugs are the same as the branded “innovator” drugs in terms of dosage, safety, strength, chemical composition, route of administration, quality, performance characteristics, and intended use (FDA, 2017a). When a generic enters the market, it tends to be priced more closely to the marginal cost of production, which often pressures the company that manufacturers the branded drug to lower the cost of that drug in
___________________
1 Many therapies benefit only some of the patients who receive them. In recent years improved diagnostic tests have become particularly valuable, especially in oncology, for providing insights, based on an individual’s genomic makeup and other biomarkers of his or her disease, into whether a particular therapy is likely to be of benefit (NASEM, 2016). Three issues may emerge in the future regarding these predictive diagnostics. The first issue involves the incentives of third-party payers to adequately compensate for diagnostics, which are often far more expensive to develop and apply than traditional laboratory tests. The second issue may arise from the increased use of companion diagnostics for rare diseases that affect only subpopulations. Third, as diagnostics advance the goal of precision medicine, the logical result will be that a given drug is prescribed for fewer patients.
order to remain competitive (Berndt and Aitken, 2011; Frank, 2007; GPhA, 2015; Grabowski et al., 2014; Greene et al., 2016; QuintilesIMS, 2016b).
People in the United States commonly pay lower prices for generic drugs than do people of other countries (Wouters et al., 2017). Generic drugs now account for up to 90 percent of all prescriptions written in the United States (GPhA, 2015; Grabowski et al., 2016). By comparison, in the early 1980s generics accounted for less than 20 percent of all prescriptions written and many profitable branded drugs with expired patents still did not have generic competitors (Frank, 2007). Analyses show that when generic drugs enter the market, they reduce the market share of the related brand drugs (Grabowski et al., 2014). If only a single generic producer enters the market, it does not necessarily reduce prices. Typically, once a drug has reached the end of its exclusivity period, the price of the branded drug may remain about the same during the period of exclusivity, or it may even drift upward, but as the generic prices decline, they capture a major portion of the market. It may take several competing generic companies to enter the market before the prices for the drug to reach their lowest possible level based primarily on cost of production.2 Generic prices, not surprisingly, exhibit the largest reductions in markets where revenues are initially above average (Gupta et al., 2016; Olson and Wendling, 2013). Multiple producers of generic drugs also help prevent shortages should one firm ceases production. From the standpoint of ensuring ongoing production and competition in the market, mergers between competing firms that make identical or biosimilar products—either generic entrants or the original branded manufacturer—are not a desirable occurrence. The Federal Trade Commission has regularly challenged such mergers (FTC, 2017).
Two recent studies examined manufacturer entry, exit, competition, and the relationships among generic drug supply structures and inflation-adjusted prices. The first of these studies found that the median and the mean number of manufacturers was about two and four, respectively, and that the number of suppliers has been declining in recent years, due both to more exit and less entry of manufacturers (Berndt et al., 2017b). The second found that a very large portion of generic manufacturers have small portfolios consisting of less than five products, while a small number of generic manufacturers have very large portfolios with hundreds or even thousands of products (Berndt et al., 2017a). Approximately 40 percent of product markets were supplied by only a single manufacturer. The share supplied by one or two manufacturers increased over time and was larger
___________________
2 The current backlog of unapproved generics at the FDA is a hurdle to generics pushing the costs of branded products down. Although the FDA’s review times for generic drug applications have decreased since the implementation of the Generic Drug User Fee Amendment, there were 2,640 generic drug applications pending approval as of April 1, 2017 (FDA, 2017f).
among non-oral drugs but varied across therapeutic classes. Generic drug prices also increased over time, particularly after 2010, following the implementation of the Patient Protection and Affordable Care Act (ACA) and the Generic Drug User Fee Amendments. The authors concluded that generic drug markets in the United States typically involve small-revenue products and are increasingly tending toward duopoly or monopoly supply. Taken together, these findings suggest that the conventional wisdom involving generic drugs in the United States—that competition among generic manufacturers, facilitated by buying power consolidation among insurers and other purchasers, results in increasing access to safe and effective treatments for chronic disease, offsetting at least to some extent the higher prices of newly launched and existing branded drugs (Aitken et al., 2016; Duggan et al., 2008)—may be less true now than previous studies suggested.
Abrupt price increases have been a matter of concern for generics as well. When the GAO examined the price histories of 1,400 generic drugs, it found 351 cases of extraordinary price increases within a single year (GAO, 2016b) (see Figure 3-2). For example, the cost of a generic antidepressant used to treat the symptoms of obsessive-compulsive disorder increased by more than 2,000 percent in 1 year, jumping from $0.34 per capsule in the first quarter of 2013 to $8.43 per capsule in the first quarter of 2014. Also, the price of a generic nonsteroidal antiinflammatory drug that can be used to treat rheumatoid arthritis or osteoarthritis increased by more than 2,000 percent, from $0.09 per capsule in first quarter 2010 to $1.94 per capsule in the first quarter of 2011 (GAO, 2016b). In some cases the prices of generics increased because of limited competition, while in other cases it was a result of delays in the review process by the FDA (GAO, 2016b; Greene et al., 2016). The lack of therapeutically equivalent drugs in the market limits competition and may contribute to extraordinary price increases (GAO, 2009). These issues further highlight the importance of having multiple producers of generic drugs. However, a recent lawsuit brought by the attorneys general of 45 states and the District of Columbia accused 18 companies and subsidiaries of colluding to fix prices for 15 medicines (Friefeld, 2017).
Biosimilars
A biosimilar is a biological product that contains a version of the active substance of an FDA-approved “reference” product (FDA, 2017b). The first biosimilar, a relative of somatropin (a growth hormone), was approved by the European Medicines Agency in 2006 (Simoens, 2011). Since then, 28 biosimilar products have been approved in Europe (QuintilesIMS, 2017b). Estimates of the overall cost saving that the European Union will experience
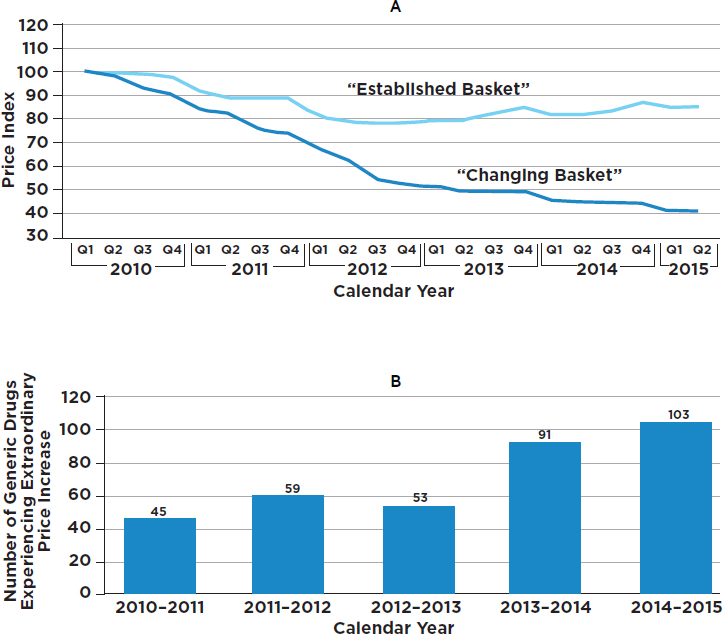
NOTES: “For the changing basket of all generic drugs, the number of drugs included in each period varies from 1,733 to 2,124. For example, the period going from the first quarter of 2010 to the second quarter of 2010 has 1,733 drugs. A total of 2,378 unique drugs were included across our study period. To be considered an established drug, a drug had to be in the Medicare Part D claims data for each quarter from the first quarter of 2009 through the second quarter of 2015 and meet certain other data reliability standards. A total of 1,441 drugs met these criteria. Due to data availability at the time we conducted our study, the second quarter of our 2015 Medicare Part D claims data is limited to data from April and May” (GAO, 2016b, p. 11).
SOURCE: GAO, 2016b, Figures 2 and 3.
by 2020 from using biosimilars range from €11.8 billion to €33.4 billion (Haustein et al., 2012).
In the United States, however, biosimilars have not yet become a major part of the drug market. There are currently only five approved biosimilars in the United States, although there are more than 60 currently under development. The first biosimilar was approved by the FDA in 2015, a version of the leukocyte growth factor filgrastim (Neupogen); this was followed by three more approvals in 2016 and one thus far in 2017. In 2009 the Biologics Price Competition and Innovation Act (BPCIA) created an abbreviated licensure pathway (351(k)) for products that are shown to be biosimilar to, or interchangeable with, a previously approved reference product.
The Congressional Budget Office (CBO) estimated that the BPCIA would result in a total cost reduction of $25 billion from 2009 to 2018. Savings to the U.S. government were projected to be $5.9 billion (CBO, 2008). An analysis by the RAND Corporation estimated that the use of biosimilar products across all therapeutic classes would result in savings between 2014 and 2024 of from $13 billion to $66 billion, depending on the amount of competition, with a best estimate of $44.2 billion (Mulcahy et al., 2014b). Among the deterrents to those wishing to bring biosimilars to market are uncertainty regarding regulatory requirements and also uncertainty about patent procedures (Hakim and Ross, 2017; Wong et al., 2017).
PRICE REGULATION
The patent law and health insurance systems in the United States are in concept similar to those in other developed countries. The United States, however, differs from most other nations with respect to the ability of the government to limit the prices of prescription drugs charged by manufacturers. While most other developed nations have governmental mechanisms for negotiating or controlling prescription drug prices, either directly or de facto (WHO, 2015), there is no nationwide regulation of drug pricing in the United States.
Table 3-1 summarizes the relevant pricing mechanisms used in five other developed nations with economies and legal structures similar to those in the United States. The tools employed in these countries include evaluating drugs using cost-effectiveness criteria and other related methods, imposing pricing limits or negotiations, and using formularies (including lists of “essential drugs,” as are discussed in Box 3-2).
TABLE 3-1
Approaches to Drug Pricing in Other Countries
| National Organization | Australia | Canada | Germany | India | United Kingdom | |
|---|---|---|---|---|---|---|
| Pharmaceutical Benefits Advisory Committee | Patented Medicine Prices Review Board | Canadian Agency for Drugs and Technologies in Health | Federal Joint Committee or the Institute for Quality and Efficiency in Health Care | National Pharmaceutical Pricing Authority | National Institute for Health and Clinical Excellence | |
| Applicability | Public payers | All payers | Public payers except in Quebec (non-cancer drugs) | All insurers | All payers | National Health Service |
| Review Criteria | Comparative effectiveness, safety, and cost-effectiveness; projected usage and overall costs to the health care system | Therapeutic innovation; comparative pricing with respect to France, Germany, Italy, Sweden, the United Kingdom, and the United States | Comparative effectiveness, safety, and cost-effectiveness; patient experiences | Comparative benefit | National List of Essential Medicines prepared on the basis of efficacy, safety, cost-effectiveness, and common diseases | Clinical effectiveness and cost-effectiveness |
| Decision | Coverage (yes, no, limited) | Price reductions or rebates | Coverage | Price setting after first year on the market | Formulary inclusion or exclusion | Coverage |
| Binding | Yes | Yes | No | Yes | Yes | Yes |
SOURCE: Adapted and expanded from Kesselheim et al., 2016.
Australia
The Pharmaceutical Benefits Scheme (PBS) was established as part of the Australian government’s broader National Medicines Policy in order to guarantee public access to (subsidized) essential medicine (PBS, 2017a). The PBS provides a list of drugs approved for coverage. To have a drug listed, its manufacturer must file an application with the Pharmaceutical Benefits Advisory Committee (PBAC), an independent body appointed by the Australian government that decides which medicines are approved and which are not (PBS, 2017b). Only those drugs on the PBS list are subsi-
dized by the Australian government. The PBAC regularly updates the list to include prescribing restrictions, maximum quantities, and price. When deciding whether to list a medicine on the PBS, the PBAC assesses the national disease burden, the medicine’s clinical effectiveness, its safety, and cost-effectiveness compared with alternative treatments. Australia uses reference pricing3 for generics and for groups of drugs with similar health and safety that can be used interchangeably. The maximum reimbursement for a medicine in a therapeutic group is based on the level of the lowest price in the approved group, and patients pay any difference between the price of the drug purchased and the reference price (Paris and Belloni, 2014).
Canada
The prices of medicines in Canada are determined by a combination of federal regulations and provincial negotiations. The price of every patented drug sold in Canada, both prescription and non-prescription, is regulated federally through the Patented Medicine Prices Review Board (PMPRB, 2017a).4 The PMPRB performs an initial review of a new drug’s price to determine if it is comparable to other products already sold in Canada. If the drug is comparable to an existing product, the price is not allowed to be greater than that of the existing drug. However, if it is not comparable, the price is allowed to be set at a point not greater than the median price in seven other industrialized countries: France, Germany, Italy, Sweden, Switzerland, the United Kingdom, and the United States. Further increases in drug prices are limited to the growth in the consumer price index (PMPRB, 2017b).
Germany
In Germany, the Act for Restructuring the Pharmaceutical Market in Statutory Health Insurance (AMNOG) established a mandatory benefit assessment of prescription drugs distributed in that country. The subsequent price negotiation process for new medicines is required to be completed within 1 year of product launch (Ruof et al., 2014). Under AMNOG, pharmaceutical companies can independently set the initial list price when they bring a new drug to market; however, they must submit a cost–benefit dossier in order for the drug to be fully reimbursed by all German insur-
___________________
3 Reference pricing involves judging the therapeutic effectiveness of drugs within a disease group and reimbursing based on the least expensive option offering comparable effectiveness.
4 The Canadian Agency for Drugs and Technologies in Health, in comparison, is responsible for making recommendations to inform coverage decisions of public drug schemes managed at the federal or the provincial level—except for Québec.
ance plans for the first 12 months. During that period, the Federal Joint Committee—the highest nongovernmental decision-making body of clinicians, hospitals, and health insurance funds in Germany—commissions a clinical comparative effectiveness review by the Institute for Quality and Efficiency in Health Care, a nongovernmental research body. Within 6 months of a drug’s introduction into the market, the Federal Joint Committee, after receiving the results of the review from the Institute for Quality and Efficiency in Health Care, will determine the new drug’s added benefits, if any, over existing drugs or treatments. The review criteria include benefits and risks for specific patient subpopulations. Each drug is given a final rating between 1 and 6, where 1 denotes “extensive benefit” and 6 means “less benefit” than an existing drug. A drug can receive different rankings for different patient subpopulations. Based on these ratings, the company then enters negotiations with the National Association of Statutory Health Insurance Funds to set the reimbursement price. One year after market launch, this reimbursement price replaces the initial list price of the drug.
Drug companies in Germany can choose to sell their products at higher prices; however, patients who want a newer, lower-ranked drug must pay the difference between the market price and the government’s set reference price. Importantly, if a drug company charges an excessive rate for a lower-ranked drug in the first year of availability, the excess revenues must be returned to payers. A drug company can opt for a drug to not be assessed, in which case the drug’s price is set through the German reference pricing system. Under the reference pricing system, the price is based on that of other drugs in the same therapeutic class, including lower-priced generic alternatives. Germany conducts more rigorous appraisals of new drugs than most other countries (Fischer et al., 2016) and has achieved significant savings in new drug spending. In 2015 Germany reported a savings of about $1 billion on new drug spending (Lauterbach et al., 2016).
India
In India, a major transition economy, the National Pharmaceutical Pricing Authority has the task of monitoring drug prices (India Department of Pharmaceuticals, 2017). By law, the authority fixes the maximum prices of items included in India’s National List of Essential Medicines. Under current regulation, manufacturers are allowed to increase prices up to 10 percent annually for medicines that are not included in the national formulary. The pricing authority also recovers overcharge amounts from manufacturers of controlled drugs; monitors drug shortages and the prices of decontrolled drugs in order to keep them at reasonable levels; and collects data on individual companies’ exports and imports, production, profitability, and market share of bulk drugs and formulations. India’s National
List of Essential Medicines is based on efficacy, safety, cost-effectiveness, and common diseases of public concern in India (WHO, 2017b).
United Kingdom
The Pharmaceutical Price Regulation Scheme, which governs drug pricing in the United Kingdom, is a voluntary arrangement between the governments of the United Kingdom and Northern Ireland and the branded pharmaceutical industry, as represented by the Association of the British Pharmaceutical Industry (Association of the British Pharmaceutical Industry, 2017). Under this regulatory scheme, which has existed in various forms since 1957, pharmaceutical prices are not directly regulated; however, if a company exceeds the profit threshold set by the government, it is given an opportunity to justify its profits and adjust the thresholds.
If the National Institute for Health and Clinical Excellence (established in 1999 to provide guidance on the clinical effectiveness and cost-effectiveness of interventions and pharmaceuticals compared with current standard practice) does not consider a new medicine to be cost-effective, it does not recommend it for use by the National Health Service (Trowman et al., 2011).
RESEARCH AND DEVELOPMENT COSTS
Estimates of the research and development costs of a new drug vary widely (Morgan et al., 2011). Decisions regarding investments in biopharmaceutical research and development depend largely on drug manufacturers’ assessment of future revenues. The greater the expected revenue from a prospective new drug, the more a drug maker will be inclined to develop it (GAO, 2009).
Spending on biopharmaceutical research and development has increased steadily over time (as addressed in the next section of this report). Revenues from the sales of prescription drugs must eventually pay for most of the costs of research and development, among other expenses, and a rise in research and development expenses will generally contribute to rising drug prices. The increase in research and development costs over time is attributable to several factors, particularly the extensiveness and cost of clinical trials, although it has been noted that the future may bring some opportunities for reducing such costs (Laurer et al., 2013).
A 2011 systematic analysis found that estimates of the cost of developing a single drug ranged from $161 million to $1.8 billion (Morgan et al., 2011). A 2016 analysis reported that the estimated cost to bring a new drug successfully to market is around $2.6 billion, with post-approval costs increasing the total to approximately $2.87 billion (DiMasi et al.,
2016). These figures are frequently cited by drug manufacturers in public and in policy discussions. A more recent analysis that considered 10 cancer drugs produced by 10 companies reported that the cost of developing a cancer drug was in the range of $157 million to $1.95 billion, with the median costs substantially lower—around $648 million, with the inclusion of opportunity costs bringing the total to $757 million (Prasad and Mailankody, 2017).
However, questions abound regarding the reliability of these studies and their estimates (Avorn, 2015; Goozner, 2017; KEI, 2014; Pitts, 2017; Wells, 2017). The basis of much of the information considered in the analyses of DiMasi and colleagues is undisclosed, and most studies have not been replicated, which raises concerns about the meaningfulness and validity of the estimates. The analysis supporting the more recent estimate from Prasad and Mailankody has been criticized for poor selection criteria. For example, critics note that their study underestimates the degree of failure in drug development by excluding larger biopharmaceutical companies that had a high percentage of cancer drug failures.
On occasion the total cost of drug development has been estimated using aggregate data on annual research and development costs reported by biopharmaceutical companies compared with the annual number of drugs approved by the FDA. Several challenges arise when using these highly aggregated data. For example, companies may conduct research and development that is not specifically related to developing novel drugs. Companies may also invest in product improvements, including the reformulation of existing drugs, as well as in analyses of the side effects of drugs already on the market. One advantage of such calculations is that they will generally take into account the large sums of money that drug companies invest in research and development on products that never reach the market. These are real costs that must be taken into account when calculating the costs of developing those products that are successful, and, indeed, publicly traded firms themselves must recognize these costs in portraying their overall financial status and also in pricing their products.
The research and development costs related to new molecular entities need to be separated from those devoted to products licensed from other firms. In the latter instance, the relevant research and development costs are reflected on the books of the licensor. Furthermore, estimates of the cost of capital that are reported in aggregated data generally do not account for the tax advantages of research and development expenditures (Riggs, 2004). In 1993, the Congressional Office of Technology Assessment estimated that the cost of research on a single drug through new drug approval was about $194 million in 1990 dollars ($363 million in 2017 dollars). The study used a marginal corporate tax rate of 34 percent, which reduced the actual cost of qualifying research and development (OTA, 1993).
The costs of abbreviated new drug applications (ANDAs) for generics have also been estimated. For oral tablets and capsules, the direct costs of ANDA applications are modest ($1 million to $5 million) compared with potential profitability (Berndt and Newhouse, 2012). Not much is known about the direct costs of obtaining ANDA approvals for infused or injected drugs.
In summary, the costs of research and development for biopharmaceutical development appear to have steadily increased in real terms over time, although it is difficult to know by exactly how much because estimates vary widely according to the analytical approach and the data sources used in making them. In a market-oriented economy, these increases in research and development costs would, over time, be expected to contribute to rising prescription drug prices.
PRODUCT PROMOTION AND DISTRIBUTION
Drug manufacturers have a direct interest in the choices made by patients and clinicians, and they have various ways to influence these choices. These include
- Discounts to PBMs and wholesalers: Manufacturers commonly sell their products at discounted prices, most importantly through the system of PBMs that is now firmly established as part of the U.S. biopharmaceutical supply chain. In concept these discounts are passed through (at least in part) from the PBMs to the consumer via the consumer’s prescription drug insurance plans—primarily through the choices of prescription drug tier and the differing copayments often associated with each drug. Greater discounts would generally be expected to lead to lower consumer copayments at the end of the supply chain. However, it is not clear that this occurs in practice.
- Marketing of products: Marketing by biopharmaceutical companies contributes to higher prescription drug expenditures through two avenues. First, studies indicate that marketing increases prescription drug use (Alpert et al., 2015; Donohue et al., 2007). Second, the costs of marketing are part of the overall cost structure of drug manufacturers and thereby place upward pressure on prices.
The exact amount that the biopharmaceutical industry spends on product promotion remains undisclosed and thus must be inferred, to the extent possible, through secondary sources of information. A recent analysis of annual financial reports and Securities and Exchange
Commission (SEC) filings of 12 large pharmaceutical companies found that between 2003 and 2015 expenditures on marketing and administration5 (a figure that includes executive pay) increased noticeably and exceeded research and development investments by up to 80 percent. Figure 3-3 displays one estimate of marketing expenditures and research and development expenditures over time.
- Direct-to-consumer advertising of pharmaceutical products: A more recent practice by pharmaceutical companies, direct-to-consumer advertising, has attracted considerable attention among those concerned with the objectivity of the process of prescribing drugs. It is noteworthy that among developed nations, the marketing of prescription drugs through direct-to-consumer advertising is legal only in the United States and New Zealand (Mackey and Liang, 2013). In recent years, direct-to-consumer advertising in the United States has grown rapidly (Wilkes et al., 2000). The Internal Revenue Code makes direct-to-consumer advertising tax deductible as a business expense, as is the case for most advertising in other industries. Recent estimates indicate that in 2016, spending on direct-to-consumer advertising was about $5.2 billion, the bulk of which was used for television promotions (Robins, 2016) (see Figure 3-4). These estimates, as compiled by Nielsen, exclude spending on Facebook, Twitter, and other digital media.
The steady growth in such advertising places increasing demands on clinicians to accommodate patient requests for advertised products that may be more costly than other treatment options (or, alternatively, to expend time explaining why an advertised medication might not be the best option for the patient). This in turn adds to the ultimate cost of drug treatment. A recent analysis of direct-to-consumer advertising concluded that these marketing efforts increased drug take-up and use—with 70 percent of the increased use arising from new patients—but also increased adherence to prescription plans (Alpert et al., 2015).
Some studies have found that direct-to-consumer advertising can increase patients’ knowledge about treatment options and may enhance the clinician–patient relationship, while others have identified effects that tend to offset these potential benefits (Lexchin, 2017; Mailankody and Prasad, 2017; Wilkes et al., 2000). In short, direct-to-consumer advertising has the potential to educate patients about conditions and their potential treatments; however, the practice may also result in unjustified demands
___________________
5 SEC filings (10-K forms) show only a blend of marketing and administration costs, thus making it difficult to isolate marketing costs as a separate item.

SOURCE: Data retrieved from Belk, 2017. See http://truecostofhealthcare.org/pharmaceutical_financial_index (accessed November 15, 2017).
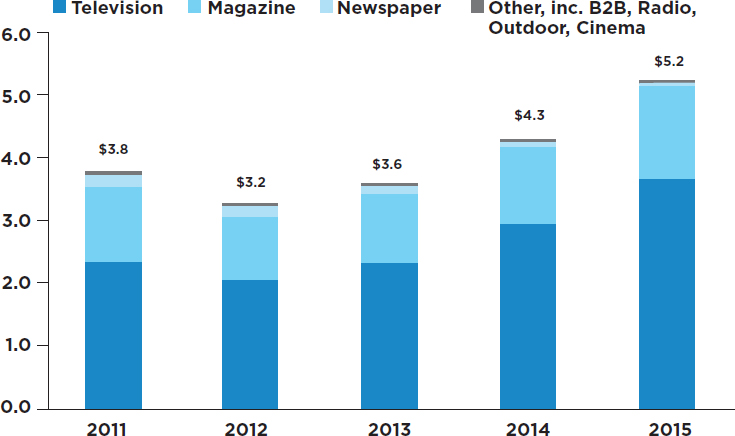
NOTE: Digital outlets not included.
SOURCES: Natalia Broshtein/STAT, from Robbins, 2016; data from Nielsen.
for expensive branded medications. The conversations triggered by this advertising may increase the pressure of clinician–patient conversations, which are already affected by short visit times, and one result may be overprescribing.
Studies of the effect of advertising on prescribing practices have shown that such advertising increases sales, reduces the underuse of some medicines needed to treat chronic conditions, and leads to some overuse of prescription drugs (Donohue et al., 2007). A randomized controlled trial to study the influence of patients’ requests for direct-to-consumer advertised antidepressants found that patients’ requests had a material effect on clinician prescribing practices for major depression and adjustment disorder (Kravitz et al., 2005). A Canadian report showed that in recent years a significant amount of money has gone toward drugs that offered “little to no therapeutic gain. This result calls into question whether doctors read journal advertisements or see sales representatives to acquire information about important medical therapies” (Lexchin, 2017, p. E724).
For more than a century there have been efforts in the United States—including legislation, regulations, and advocacy—to control the marketing and advertising of pharmaceuticals directly to consumers (Mogull, 2008). Recently, the American Medical Association called for a complete ban on direct-to-consumer advertising, arguing that the “growing proliferation of ads is driving demand for expensive treatments despite the clinical effectiveness of less costly alternatives” (AMA, 2015). The Congressional Budget Office (2011) examined the potential effects of a moratorium on direct-to-consumer advertising of new prescription drugs and concluded that:
- Drug manufacturers would probably expand their marketing to clinicians to substitute for at least some of the banned advertising to consumers.
- The number of prescriptions filled would probably decrease for some drugs, but for other drugs the number of prescriptions might be little changed, owing both to the likely substitution of other types of promotions and to other factors that influence a drug’s reach in the prescription drug market.
- Any change in prescription drug prices would depend on changes in demand; however, prices for new brand drugs that normally would be part of a direct-to-consumer advertising campaign could increase, since sales would be reduced.
- A moratorium could affect public health. The exact result would depend on whether the benefits of fewer unexpected adverse health events were greater than the health costs of possibly reduced use of new and effective drugs.
While the results of studies of the effects of direct-to-consumer advertising are somewhat inconclusive or at least mixed, drug advertisements remain pervasive and influence the manner in which clinicians prescribe. Because advertising is demonstrably effective in stimulating consumer demand for branded drugs and adds to the cost of doing business, such direct-to-consumer advertising likely contributes to the nation’s high prescription drug costs.
The FDA regulates the content of this advertising, seeking to ensure a fair balance in describing benefits and risks and making certain that the risks are included in a prominent statement (Ventola, 2011). Although proposals exist to ban direct-to-consumer advertising of drugs, the constitutional protection of free speech in the United States may constrain such efforts.6 The U.S. Supreme Court has regularly ruled that commercial speech is protected by the First Amendment.
Marketing practices such as direct-to-consumer advertising aside, there are several other ways that manufacturers influence the debates and discussions in the biopharmaceutical sector, some of which are described in Box 3-3.
- Direct rebates to consumers: Another mechanism used by pharmaceutical manufacturers to affect the choices of patients and prescribers is the provision of direct payments to patients upon proof that they are actually using the specific drug. These payments have two key features. First, they almost universally are
___________________
6 An often compared scenario is the federal legislation that banned advertising of tobacco on television and radio beginning in 1971. In 1967, the Federal Communications Commission ruled that the “fairness doctrine” be applied to cigarette advertising, which meant that television stations that broadcasted tobacco advertisements were required to give equal time to showing anti-smoking messages, during prime time as well as during children’s programs, leading up to the Public Health Cigarette Smoking Act of 1969, which banned cigarette advertisements on American radio and television.
After the ban, the tobacco industry increased advertising in other media, but the total volume of advertising by the industry decreased. In parallel, broadcast media no longer were compelled to run anti-smoking messages, and their removal may have contributed at least briefly to increase smoking rates. The anti-smoking advertisements were mandated by the Federal Communications Commission in response to the strong evidence of harm caused by tobacco, a unique situation.
The ban also appears to have reduced competition in the industry, allowing the firms selling cigarettes prior to the ban (the incumbent firms) to maintain higher prices than they would have if they had been challenged by market entrants. Without the ban, market entrants might have attracted consumers away from the incumbent firms using television and radio advertising. The financial advantage to incumbent firms may explain why the tobacco industry did not challenge the advertising bans (Eckard, 1991). The reduced cost of advertising may also have increased tobacco industry profits, a situation that might be repeated in the biopharmaceutical sector.
employed by makers of on-patent drugs—often in situations where competition exists from either generic or other branded drugs. Second, they are generally directed at patients with prescription drug insurance plans, using such language as “if you need help with your copayments.” Such rebates have the effect of counteracting higher-tier (larger) copayments set by PBMs and health insurance plans, thereby increasing annual insurance premiums for all enrollees in prescription drug plans but reducing the drug cost to the individuals receiving the rebate. A comparison of Figures 2-5
and 2-6 illustrates these mechanisms. One recent analysis estimated that copay coupons increase branded drug sales by 60 percent or more, almost entirely by reducing the sales of generic competitors, and that branded drug manufacturers receive a return of between four-to-one and six-to-one on every dollar spent on copay coupons (Dafny et al., 2016a). Analyses have also concluded that copay coupons increase costs for all enrollees in prescription drug insurance plans (Dafny et al., 2016a; Ross and Kesselheim, 2013).
IMPORTATION OF MEDICINES
One strategy that has long been advocated as a way of reducing prescription drug prices and countering drug shortages (see the section on drug shortages) in the United States is to import prescription drugs—especially generics and biosimilars—from other countries. The rationale is that importing lower-cost drugs from other countries with high-quality production systems (and, potentially, government limits on price increases) would cause U.S. manufacturers to be faced with greater competition and encourage them to reduce prices.
A related strategy is “reimportation,” or having U.S. wholesalers and pharmacies import and sell branded drugs that were produced in the United States but sold in other countries where prices are lower, as long as the FDA has approved a version of the same drug for domestic use.7 Essentially, the goal of reimportation is to negate drug manufacturers’ differential pricing across countries—and, in particular, the pattern of charging more in the United States than in other countries for the same drug (Outterson 2005). Such programs could in principle also be established by state or local governments.
The importing and the reimporting of prescription drugs have been perennial proposals in the U.S. Congress over the past two decades. A number of states and localities have experimented with pilot programs, which have generally encountered legal challenges. Despite the longstanding interest, efforts to legalize the practice have not been successful.
The Food, Drug, and Cosmetics Act (FDCA) prohibits the importation of prescription drugs made in the United States by anyone other than the manufacturer—with the exception of drugs approved by the secretary of the U.S. Department of Health and Human Services (HHS) for emergency care. Another legal obstacle is that it is nearly impossible for drugs made for non-U.S. markets to satisfy the FDCA’s requirements relating to drug approval and labeling (CRS, 2008; Terry, 2004).
Importation and reimportation run the risk of enforcement actions for introducing “misbranded” drugs into U.S. markets. The federal Controlled Substances Act also bears on reimportation in that it prohibits the unlawful distribution of prescription drugs, such as narcotics and opioids, that meet the statutory criteria for controlled substances (CRS, 2008).
In 2000, Congress passed the Medicine Equity and Drug Safety Act
___________________
7 “Personal reimportation” proposals focus on making it easier for individual U.S. consumers to buy and import drugs from other countries. Personal reimportation is not permitted by law, but the U.S. Department of Health and Human Services has historically exercised its discretion not to enforce this prohibition against individuals bringing medications into the United States for personal use (Reichertz and Friend, 2000). This report’s focus is on broader-scale reimportation proposals, which have greater potential for population-level impact.
that authorized the FDA to allow the reimportation of prescription drugs from a specified group of countries. The U.S. Congress again authorized reimportation in the Medicare Modernization Act, this time narrowing the list of acceptable countries to Canada. But neither act was implemented, due to opposition by the HHS secretary. Both statutes required the secretary to certify that reimported drugs would be safe and would significantly reduce costs. No secretary has yet been prepared to do so. Thus, importation and reimportation remain prohibited. Several subsequent legislative proposals have failed to clear these and other obstacles (Bluth, 2017).
Historically, the FDA has opposed reimportation out of concerns about its ability to ensure a safe drug supply (Bhosle and Balkrishnan, 2007; Terry, 2004). Although reimportation discussions often assume that the drugs would be imported from Canada, if a sufficient supply could not be obtained there it could become necessary to import from countries with a less robust record of preventing counterfeit, contaminated, expired, and mislabeled drugs from reaching the market. Moreover, drugs may be falsely labeled as originating in the United States or a Canadian pharmacy (Bhosle and Balkrishnan, 2007). FDA commissioners have consistently expressed skepticism about the agency’s ability, with its current financial and technological resources, to ensure the safety and authenticity of a much larger volume of imported drugs (Bhosle and Balkrishnan, 2007). Others note, however, a lack of evidence that Canadian drugs are less safe or that concerns about adulteration and other problems are unique to imported drugs (Kamath and McKibbin, 2003; Outterson, 2005). The proponents of importation have argued for further pilot studies of controlled importation systems, noting that the reimportation ban values drug safety absolutely, at the expense of financial access, and arguing that safety concerns have been overstated.
Even if importation or reimportation, or both, were allowed, it is not clear how much they would reduce drug costs for U.S. consumers. The outcome would depend largely on (1) the countries from which drugs may be imported or reimported, and (2) the strategic responses of U.S. drug manufacturers. The CBO (2003) has estimated that allowing reimportation from 25 countries would save $40 billion over 10 years; however, other research has concluded that the savings would be considerably smaller, about $1.7 billion annually (Danzon et al., 2011).
A key question is how large a supply of drugs Canada and other approved countries would make available for export back to the United States (or, in the case of generics, allow them to be imported by the United States at lower prices). The CBO has concluded that the savings would not be substantial if reimportation were limited to Canada because drug companies probably would not increase their Canadian sales enough to allow a significant propor-
tion of American-made drugs to be reimported from Canada (Kaiser Health News, 2009).
It is also possible that manufacturers could penalize countries and firms that exported products back to the United States or that imported generics and biosimilars to the U.S. market. Firms could, for example, raise the prices of drugs sold in Canada, or penalize wholesalers that reimported drugs by raising the prices of the drugs they sell to those particular wholesalers in the United States. There is anecdotal evidence of penalizing behavior on the part of U.S. manufacturers: in 2004, GlaxoSmithKline and Pfizer announced that they would limit sales of their drugs to Canadian pharmacies that resold them to individual U.S. consumers (CRS, 2008). A larger question is whether importation and reimportation would spur manufacturers to reduce their investment in research and development.
DRUG SHORTAGES
In recent years there have been numerous high-profile reports of inadequate supplies of generic drugs that have served as the standard of care for some diseases. For example, shortages have been reported for two critical cancer drugs, Doxil and Methotrexate, a medication used as backbone therapy to treat pediatric cancer (Harris, 2012); various antibiotics, including doxycycline (Stone, 2015); and saline bags, which are used throughout inpatient and outpatient treatment (McGinley, 2017).
Although the number of new drug shortages has declined since 2011, prominent shortages exist among generic injectables and other drugs for cancer and cardiovascular conditions (ASHP, 2017a; GAO, 2014, 2016a), and drug shortages have been known to lead to adverse events and even increased patient morbidity and mortality (Duke et al., 2011; Gu et al., 2011; Kaakeh et al., 2011; Kaiser, 2011; McKenna, 2011). The more constrained supply of such drugs has also led to higher prices for these drugs (GAO, 2014; IOM, 2013).
Shortages, threatened and actual, often result from lapses in manufacturing quality (Fox et al., 2014; GAO, 2016a; Stomberg, 2016). For example, the immediate precipitating factors behind the shortages reported since 2009 (largely for infused and injectable drugs) include a lack of high-quality manufacturing processes and facilities and a lack of necessary compounds and raw materials (GAO, 2014; Pew, 2017; Stomberg, 2016; Woodcock and Wosinska, 2013). The lapses in manufacturing quality and the shortages in the of necessary or adequately manufactured raw materials that can lead to supply interruptions of certain drugs and other products regulated by the FDA are not new, but appear to be more frequently reported in recent years. For example, in 2008, the FDA reported that at least 81 deaths and 785 serious injuries were thought to be linked to a
raw heparin ingredient imported from China (FDA, 2012). This led to the withdrawal of the product from the U.S. market for a period of time and consequently there was an inadequate supply to meet American demand.
Yet, according to a recent report from the American Society of Health-System Pharmacists, the immediate causes for more than one-half of drug shortages reported in 2016 were unknown (ASHP, 2017b). In some circumstances, unexpected consumer demand or an outbreak of a rare illness can contribute to drug shortages (ASPE, 2011; Fox et al., 2009; GAO, 2014; IOM, 2013; Pew, 2017). In addition, a federal report noted that class-wide shortages in 2011 were likely due to a rapid and sizeable increase in the scope and volume of products produced without a corresponding increase in overall manufacturing capacity (ASPE, 2011). The constrained supply of these drugs and the high costs of entry for manufacturers willing and able to produce these molecules for sale in the U.S. market also contribute to threatened and actual drug shortages (ASPE, 2011; Berndt et al., 2017a,b; Fox et al., 2009; IOM, 2013). The FDA response to periodic drug shortages has largely been to either pull or push more manufacturers into supplying U.S. demand for these products.
The growing trend to outsource drug manufacturing and to source base ingredients from non-U.S.-based manufacturing facilities, along with the highly publicized incident of adulterated heparin manufactured in China that evaded inspection by a resource-constrained FDA (U.S. Congress, 2008), led to key aspects of the Generic Drug User Fee Amendment (GDUFA), first enacted in 2012 (Conti and Berndt, 2017a). Specifically, GDUFA funded the FDA’s redesign of its inspection program and the associated user fee schedule to meet these new challenges.
More recently, under the Safety and Innovation Act of 2012, the FDA required drug manufacturers to provide early notification of any manufacturing interruptions or production changes that could lead to a supply disruption or the discontinuation of a product. Subsequently, the FDA improved its efforts to prevent shortages by expediting application reviews and inspections, exercising enforcement discretion in relevant cases, and helping manufacturers respond to quality control issues in drug manufacturing (Chen et al., 2016; GAO, 2016a).
WASTE AND COST DUE TO UNUSED DRUGS IN THE SUPPLY CHAIN
Every year drugs worth billions of dollars that have been purchased by health care organizations (e.g., retail pharmacies, hospitals, nursing homes) and patients are discarded. Some of this waste in the system could be eliminated by changing the way drugs are packaged and labeled. For example, vials of infused drugs are often available only in a single dose size that is
sufficient to treat a physically large patient. As a result, the remaining drug must be discarded when a smaller patient is treated. Because 18 of the top 20 infused cancer drugs are sold in just one or two vial sizes, 10 percent of the purchased drug amount is discarded on average (Bach et al., 2016). Manufacturers propose dose sizes for marketing, and the FDA only reviews the request for safety considerations (FDA, 2015). However, in Europe, where governments play a more active role than the United States does in drug pricing and distribution, many of these medicines are distributed in smaller vial sizes, reducing the potential for waste.
Many medicines are also discarded because of expiration dates (Allen, 2017). Since 1979 the FDA has required drug manufacturers to provide evidence of product stability, by subjecting drugs to various environmental variables such as temperature, humidity, and light, but there are no requirements for long-term testing. Pharmacies routinely discard stocked drugs when they reach their expiration date, but many drugs, if stored properly, are stable long beyond the expiration date on the label (Cantrell et al., 2012, 2017; Lyon et al., 2006). The strongest evidence comes from the FDA’s Shelf Life Extension Program (SLEP) (FDA, 2017c), which is funded by the U.S. Department of Defense to support the maintenance of its stockpiled drugs, worth billions of dollars. In a study of 122 different medication products, nearly 90 percent met the requirements for an extension; the average additional extension length by SLEP was 5.5 years, and some lots were extended by more than 20 years (Lyon et al., 2006).
Extending shelf life could not only reduce waste in the system, but also address shortages. The FDA recently posted updated expiration dates for batches of several different injectable drugs to help address ongoing critical shortages of these drugs used in critical care (FDA, 2017d). The American Medical Association and other entities have called for routinely collecting more data on long-term stability and revising expiration dates as appropriate (Diven et al., 2015). An independent organization could conduct more testing similar to that done by the FDA extension program. Information from the extension program also could be applied to properly stored medications.
Drugs worth billions of dollars are discarded each year by nursing homes and other long-term care facilities when they are no longer needed by residents (Allen, 2017; Coggins, 2016). A few states and nonprofit organizations have set up programs to collect, sort, and redistribute these unused drugs to reduce waste and costs to patients. However, in many areas no such programs exist (and in some cases are even illegal), so valuable drugs are simply discarded.
INSURANCE DESIGN
A key factor affecting the affordability of health care for individuals and families is whether a patient has health insurance. After the implementation of the ACA, the number of people with health insurance increased substantially, but approximately 10 percent of the population under age 65 has no health insurance—and hence no coverage for prescription drugs. Furthermore, not all of those with health insurance have insurance coverage for prescription drugs. This latter circumstance applies to both the under-65 population and those on Medicare. Fee-for-service Medicare helps cover the cost of prescription drugs for people who enroll in a Part D drug plan (see Figure 3-5), but enrollment is voluntary and only 42 million of the 57 million Medicare beneficiaries have Part D coverage (KFF, 2017b). However, of the remainder, some have drug coverage through employers, the U.S. Department of Veterans Affairs, and other “creditable” sources (those that offer coverage as good as is provided by Part D), but a small share (about 12 percent) of Medicare beneficiaries lack a creditable source of drug coverage (MedPAC, 2017a). As of 2017, 99 percent of covered

NOTES: Some amounts rounded to nearest dollar. 1Amount corresponds to the estimated catastrophic coverage limit for non-low-income subsidy (LIS) enrollees ($7,425 for LIS enrollees), which corresponds to a true out-of-pocket spending of $4,950, the amount used to determine when an enrollee reaches the catastrophic coverage threshold in 2017.
SOURCE: KFF, 2017.
employees worked for a firm whose largest health plan covered prescription drugs (KFF and Health Research & Educational Trust, 2017).
Recent changes in insurance design in the United States reflect the rising costs of not only drugs but all sectors of health care (Consumer Reports, 2016). As the costs of health care have risen, employers and insurers have modified benefit designs as a way to keep premiums as low as possible, with the goal of balancing cost and access. The escalating list prices of many branded drugs, especially specialty drugs and those that lack a competitor, have been a particular challenge in recent years.
As a result, even among those with insurance benefits, the out-of-pocket costs for premiums, deductibles, and copays can be substantial, and the design of the coverage and cost sharing can significantly affect the financial burden arising from prescription drug spending. Studies have found dramatic reductions in coverage generosity and shifts to percentage-based cost sharing for high-priced drugs over time (Doshi et al., 2016a; Dusetzina, 2016; Jung et al., 2016; Polinski et al., 2009; Yazdany et al., 2015), limiting options for patients to obtain plans that provide generous coverage for drugs. The specifics of pharmacy benefit design have the potential to be an important public health tool for improving patient treatment and adherence (Goldman et al., 2007) and can have a major effect on access to prescription medications (Delbanco et al., 2016).
The effects of high out-of-pocket spending can be significant for patients and their families. Increased cost sharing can reduce patient uptake and adherence to treatments, including specialty drugs (Alexander, 2003; Doshi et al., 2016a,b; Dusetzina et al., 2014; Fischer et al., 2011; KFF, 2015; Olszewski et al., 2017; RAND Health, 2006; Streeter et al., 2011; Winn et al., 2016). Nearly one-quarter of the Americans who participated in a 2015 survey reported that they had difficulty affording their prescription medicines. And nearly one-quarter reported that they or a family member had not filled a prescription that they had been provided, had skipped doses, or had reduced their dosage because of cost (KFF, 2015). One study of commercially insured adults with chronic myelogenous leukemia found that having higher out-of-pocket costs reduced patient adherence to therapy by 42 percent and increased the discontinuation of therapy by 70 percent (Dusetzina et al., 2013). A 2014 study on primary care found that approximately 31 percent of patients did not fill their prescriptions within the first 9 months after receiving them from a doctor. Additionally, the study found that patients with higher copayment fees, recent hospitalizations, severe comorbid conditions, or some combination of these three factors were less likely to fill their prescriptions (Tamblyn et al., 2014). Various studies confirm that poor adherence leads to negative clinical outcomes and increased health care costs (e.g., Roebuck et al., 2011).
Even for insured patients, the use of prescription drugs often entails a large out-of-pocket expense because of the high coinsurance rates that often apply to expensive drugs, particularly if the patients use specialty drugs or multiple high-cost brand-name drugs. For example, traditional Medicare currently places no upper limit on the total amount an individual may end up spending on cost sharing for Medicare-covered services. For services offered under Medicare Part B, including clinician-administered drugs, the beneficiaries (or their supplemental insurance plans) are responsible for 20 percent of the cost (MedPAC, 2016), and there are no catastrophic coverage limits. In 2014, virtually all Part D formularies required coinsurance of between 25 and 33 percent for cancer drugs, the maximum allowed by the Centers for Medicare & Medicaid Services (CMS) (Dusetzina and Keating, 2015). This can translate to hundreds or even thousands of dollars annually in out-of-pocket costs for higher-cost medications.
Individuals with employer-sponsored or other types of private health insurance also face challenges with prescription drug costs. As noted above, in response to increasing costs across all sectors of health care, insurance companies have raised deductibles, increased monthly premiums, imposed or increased copays and coinsurance, and transferred high-cost drugs to more expensive formulary tiers (Claxton et al., 2017; Consumer Reports, 2016). Among employer-sponsored plans, deductibles grew from 4 percent of cost-sharing payments in 2004 to 24 percent in 2014; coinsurance increased from 3 to 20 percent over that same period (Cox, 2016). A patient’s costs often depend on the tiered structure by which many health plans organize the drugs that are covered by their formularies.
Private health insurance plans have been moving to three- or four-tier coinsurance or copayment structures that require a smaller degree of cost sharing for generics and a greater degree for higher-cost drugs especially when there are therapeutically equivalent options (KFF, 2014). Plan tiers often include preferred, non-preferred, and generic drugs, and each tier of drugs can represent a different level of cost sharing or class of drugs, such as specialty drugs (KFF and Health Research & Educational Trust, 2017). However, every plan, whether Part D or an employer-sponsored pharmacy benefit, has an exception process that permits coverage of a drug not on formulary or reduce out-of-pocket cost if a physician provides information about side effects the patient has experienced from a lower-tiered drug or offers another medical reason for switching.
As noted in Box 2-1, most large employers self-finance their health insurance contributions for their employees and hence have a direct and significant interest in controlling health care costs. As of 2017, 91 percent of employees covered by employer-sponsored insurance plans were in a plan with tiered cost sharing (KFF and Health Research & Educational Trust, 2017). The ubiquity of such plans can influence how much individuals cov-
ered by specific employer-sponsored plans pay due to the variation in cost sharing. The Kaiser/Health Research & Educational Trust 2017 Employer Health Benefits Survey of workers covered by employer-sponsored plans found that, among those in plans with at least three tiers of cost sharing, the average copayment per drug was $11 for the first tier and $110 for the fourth tier. The average coinsurance was 17 percent for first-tier drugs and 38 percent for third-tier drugs. Specialty drug tiers tend to drive up cost sharing even further, with an average copayment of $101 and an average coinsurance rate of 27 percent for drugs on a specialty tier. In addition to copayments and coinsurance, health plans can apply an additional deductible to drugs that is separate from the general annual deductible. In 2017, 15 percent of workers with prescription drug coverage had to meet a prescription drug–only deductible (KFF and Health Research & Educational Trust, 2017).
Health plan decisions regarding which drugs to include in their formularies—and in which tiers—also reflect the influence of PBMs, whose negotiations often occur with minimal transparency or data on rebate amounts, raising concerns about their impact on patients’ out-of-pocket spending (Health Affairs, 2017b). However, Part D plans do enable consumers to determine and compare the out-of-pocket costs of a drug in the “preferred pharmacy network,” “non-preferred networks,” and mail-order services.
“High-deductible health plans” are also becoming more common in the U.S. insurance marketplace as health care costs rise (Claxton et al., 2016). These plans require a higher deductible than most health plans, in exchange for a lower monthly premium. High-deductible health plans require consumers to cover 100 percent of their health care costs up to a certain amount—the deductible—at which point their insurance coverage and other cost-sharing arrangements begin. In 2016, nearly 30 percent of individuals in employer-sponsored plans were enrolled in a high-deductible health plan (Claxton et al., 2016). Recent work has begun to explore the clinical and economic benefits of high-deductible plans in the long run (Fronstin et al., 2013).
Out-of-Pocket Spending and Specialty Drug Access
Many oral drugs used to treat complex conditions such as HIV, multiple sclerosis, rheumatoid arthritis, cancer, and hepatitis C are costly, and the increasing use of deductibles and coinsurance in the pharmacy benefit offered by insurance plans may lead to significant financial hardship for patients needing treatment. Medicare beneficiaries are exposed to high costs in two primary ways. First, enrollees who take drugs covered on a Part D plan’s specialty tier face coinsurance rates of between 25 percent
and 33 percent of the drug’s total price during the initial coverage phase. In 2017 the Part D standard benefit has a $400 deductible and 25 percent coinsurance up to an initial coverage limit of $3,700 in total drug costs, followed by a coverage gap (see Figure 3-5). During the gap, enrollees are responsible for a larger share of their total drug costs than in the initial coverage period, until their total out-of-pocket spending in 2017 reaches $4,950. After enrollees reach the catastrophic coverage threshold, Medicare pays for most (80 percent) of their drug costs, plans pay 15 percent, and enrollees pay 5 percent of total drug costs. Second, even patients who reach the catastrophic coverage threshold of Medicare Part D can be exposed to high costs because the threshold is not a hard cap on out-of-pocket costs. For medications costing tens of thousands of dollars or more per year, patients can spend more out of pocket during the catastrophic phase than in the other benefit phases combined (Hoadley, 2015). A recent analysis found that 3.6 million Medicare beneficiaries had total drug spending above the Part D catastrophic threshold in 2015, and of those, one million incurred out-of-pocket drug costs above the threshold (KFF, 2017a).
Two examples illustrate the extent to which Part D enrollees can be exposed to serious financial risk, despite the Part D benefit’s catastrophic coverage, when the underlying price of the drug they take is very high. For Harvoni, a breakthrough treatment for hepatitis C, a patient enrolled in Part D in 2016 faced total out-of-pocket costs of $7,153 for a course of treatment, but 61 percent of this total was incurred in the catastrophic coverage phase. For Revlimid, a cancer drug, a patient enrolled in Part D in 2016 faced total annual out-of-pocket costs of $11,538 for this drug alone in 2016, 76 percent of which was in the catastrophic coverage phase of the benefit. (The price for Revlimid has since increased dramatically, to more than $18,000 per fill; thus, in the catastrophic phase under Part D, enrollees will pay more than $900 per month for this drug [Court, 2017].)
One way to strengthen financial protections for Medicare beneficiaries with very high drug costs would be to eliminate enrollees’ cost sharing above the catastrophic coverage threshold, thereby making the current catastrophic coverage threshold an absolute limit on out-of-pocket spending under Part D. This proposal has been recommended by the Medicare Payment Advisory Commission (MedPAC, 2016). To mitigate the concern that pharmaceutical companies might respond by simply raising their list prices, one strategy might be to increase the share of total costs that Part D plan sponsors pay in the catastrophic coverage phase of the benefit (up from the current 15 percent), giving them a stronger financial incentive to negotiate larger rebates for higher-priced drugs and to take more steps to manage the use of these drugs by their enrollees, which could produce savings for enrollees, Medicare, and the plans themselves.
PATIENT ASSISTANCE PROGRAMS
Patient assistance programs supported by drug manufacturers serve to lower patients’ out-of-pocket spending by covering the cost of all or part of their out-of-pocket expenses when they buy brand-name medications. This practice may bring the price that patients pay for branded medications closer to—and in some cases lower than—the price of generic alternatives, but it does not change the cost to the insurer. In fact, such practices serve to increase costs to insurers and therefore, the premiums charged by the insurer. These practices also lessen the insurer’s ability to price discriminate (through the use of tiers in the formulary), as patients with access to these programs will often opt for branded products over generics and, more generally, choose drugs not “preferred” by the plan (Ubel and Bach, 2016).
The popularity of patient assistance programs among both patients and manufacturers has increased over time (Daubresse et al., 2017; Ross and Kesselheim, 2013). Assistance programs are delivered through a variety of mechanisms, including coupons, drug savings cards, manufacturer assistance programs (provided through the drug maker), access networks that create disease-specific funds, and disease-focused foundation programs. Payments are generally distributed via clinicians’ offices or, increasingly, directly to the patient through the mail or online (Dafny et al., 2016b). Support can include providing medications or payments directly to individuals. Eligibility for support from these sources varies by insurance status and income. Some types of copayment assistance are not allowed, including the use of manufacturer coupons to pay for drugs obtained through Medicare Part D benefits.
While helpful in some ways, patient assistance programs encourage patients to use higher-cost branded products, since generic manufacturers do not typically offer assistance programs. Patients with very high deductibles or with high coinsurance requirements may find it difficult to pay the out-of-pocket costs to obtain high-priced drugs. In such cases, patients may need to access assistance programs in order to offset the out-of-pocket costs of starting and adhering to therapy, regardless of their insurance status. Each program is a unique, unregulated, private offering by a pharmaceutical company for an individual product. The application process can be onerous for patients and clinicians, with a high probability of rejection, commonly based on patient income level and insurance coverage. There is little information available to evaluate the impact of patient assistance programs so few studies have examined the proportion of patients served, the extent of aid provided, the criteria for qualifying for aid, and the estimated financial cost to society (Felder et al., 2011).
Drug manufacturers tend to use coupons to promote the use of branded expensive products when less expensive alternatives are available (Dafny
et al., 2016b; Ross and Kesselheim, 2013). One analysis estimated that copay coupons increased branded drug sales by 60 percent or more, almost entirely by reducing the sales of generic competitors, and that they had the potential to undermine the efforts of prescription drug insurance plans (Dafny et al., 2016a). Federal policies prohibit the use of manufacturer coupons in paying for medications paid for by Medicare Part D because it is considered a violation of anti-kickback statues and it raises costs to the government (OIG, 2014).
FEDERAL DISCOUNT PROGRAMS
Medicaid Drug Rebate Program
The U.S. Congress created the Medicaid Drug Rebate Program (MDRP), which went into effect in 1991, resulting from the Omnibus Budget Reconciliation Act of 1990 in an attempt to address the rising cost of prescription drugs in the Medicaid program. In the MDRP, the drug manufacturer enters into a rebate agreement with the HHS secretary in return for Medicaid coverage of all products made by this manufacturer, as well as payments for covered outpatient drugs provided through Medicare Part B. This has essentially created an open formulary in Medicaid. CMS reports that about 600 manufacturers have entered into such an agreement (CMS, 2017a).
Unlike most rebates for prescription drug spending, the rebates obtained through the MDRP are not negotiated, but are defined by statute. However, many components used to calculate the rebate are proprietary, and as a result, it is difficult to calculate exactly how much Medicaid spends on a particular drug. This contributes to the lack of transparency surrounding drug pricing. Statutory rebates are set by the U.S. Congress and enacted into law, and there have been changes over time. States are free to negotiate supplemental rebates on top of the statutory rebates.
The basic rebate calculation for single-source drugs and innovator multiple-source drugs8 is set by statute and is set separately for non-innovator, multiple-source drugs. For single-source and innovator multiple-source drugs, the unit rebate amount is equal to the greater of either the product of Average Manufacturer Price (AMP) times 23.1 percent or the difference between AMP and the Best Price (defined as the lowest
___________________
8 “Innovator drugs” include both single-source (typically a brand-name product that has no available generic versions) and multiple-source (typically a brand-name product that has available generic versions) products. “Non-innovators” are typically generic versions of multiple-source drugs (OIG, 2009). Statute sets different rebate percentages for certain types of single-source and innovator multiple-source drugs: clotting factors and drugs approved by the FDA exclusively for pediatric indications.
price the manufacturer charges any wholesaler, health maintenance organization, retailer, health care provider, government entity, or nonprofit organization in the United States during that rebate period). For non-innovator, multiple-source drugs, the unit rebate amount is equal to the product of AMP times 0.13. The rebate on innovator drugs includes an adjustment to account for price inflation; however, this adjustment is not included in the rebates for non-innovator drugs. There are some important exceptions to the “best price” provision, including prices charged in the 340B program to the U.S. Department of Veterans Affairs, the U.S. Department of Defense, Medicare Part D, and Indian Health Service. This seemingly minor part of the MDRP has a major implication: outside of these exceptions, manufacturers are very reluctant to provide rebates for single-source or innovator multiple-source drugs large enough to trigger the “best price” provision because it would create a lower price for the entire Medicaid program (Health Affairs, 2017a).
Rebates are paid by drug manufacturers on a quarterly basis to states and are shared between the states and the federal government. Prior to the ACA, rebates through the MDRP were only available for drugs provided in fee-for-service settings. Under the ACA, drugs provided in managed care settings are also eligible for rebates and as a result, states have increasingly been providing the Medicaid prescription drug benefit through managed care.
In large part due to the market entry of very expensive hepatitis C drugs, Medicaid expended $57 billion on prescription drugs in 2015, compared to $42 billion in the previous year (Health Affairs, 2017a). States have been left vulnerable to the high costs of branded drugs that have little competition (McConnell and Chernew, 2017). The National Association of Medicaid Directors has called for expanding the tools that states can use to design and manage Medicaid’s optional prescription drug benefits, including providing states with the flexibility to exclude some FDA-approved drugs from coverage (NAMD, 2017). Recently, Massachusetts submitted an amendment to its 1115 demonstration waiver to CMS that would allow the state to have a closed formulary.9 However, in response to this waiver request, some advocates have emphasized the importance of specifying exclusion criteria in order to ensure that patients with serious conditions on Medicaid are not denied needed effective treatments for their conditions.
The 340B Program
Prior to the implementation of the MDRP in 1991, manufacturers often provided discounts on their drugs to safety net providers. However,
___________________
9 Commonwealth of Massachusetts, MassHealth Section 1115 Demonstration Amendment Request, September 8, 2017. The waiver request is pending as of November 2017.
after the establishment of the MDRP, there was concern that manufacturers would be less willing to provide additional discounts to providers serving largely uninsured or underinsured populations. The U.S. Congress addressed this potential unintended effect of the MDRP with the 340B program, a drug discount program named after the section10 in the law that created it (Health Affairs, 2017a). The stated goal of the 340B program is to enable these providers “to stretch scarce federal resources as far as possible, reaching more eligible patients and providing more comprehensive services” (HRSA, 2017).
Section 340B requires certain drug manufacturers to provide outpatient drugs to qualified medical care providers, called “covered entities,” at prices not higher than Medicaid is able to obtain, net of rebates (GAO, 2011; Health Affairs, 2014; HRSA, 2015). Covered entities can seek additional rebates on top of the 340B discount. 340B discounts and potential additional price concessions are not included in the Medicaid best price (Health Affairs, 2017c). The law instructs the HHS to enter into a pharmaceutical pricing agreement (PPA) with drug manufacturers as a stipulation for their drugs to be covered under Medicaid. If a drug manufacturer signs a PPA, it agrees that the prices charged for covered outpatient drugs to covered entities will not exceed 340B ceiling prices as defined by statute. The Health Resources and Services Administration (HRSA) calculates the ceiling prices quarterly using pricing data reported to CMS. The 340B ceiling price is calculated by subtracting the Unit Rebate Amount from the AMP. In practice, this results in the 340B price being about 20 to 50 percent off the drug’s AMP. Drugs included in the 340B program generally consist of outpatient prescription drugs and drugs administered by clinicians in an outpatient setting, excluding vaccines.
HRSA administers the 340B program and is responsible for the oversight of various stakeholders, including covered entities and pharmaceutical companies. The ACA expanded the types of covered entities eligible to participate in the 340B program, including critical-access hospitals, rural referral centers, sole community hospitals, and freestanding cancer centers. Furthermore, in 2010, HRSA allowed 340B entities to sign agreements with more than one outside pharmacy—known as contract pharmacies—to provide the covered drugs. Contract pharmacies are employed by some hospitals and clinics to expand services outside of hospital walls (HRSA, 2010).
By design, 340B program participation provides qualified entities the opportunity to generate revenue from administering and dispensing prescription drugs, financed by pharmaceutical manufacturers, insurers, and paying patients (Conti and Bach, 2014). The program does not require enti-
___________________
10 Section 602 of Public Law 102-585, the Veterans Health Care Act of 1992 enacted section 340B of the Public Health Service Act (PHSA) Limitation on Prices of Drugs Purchased by Covered Entities, codified at 42 U.S.C. 256b.
ties to pass the drug discounts along to the patients they treat in the form of lower out-of-pocket costs, nor does it require passing those discounts in the form of lower reimbursements to the insurance plans that cover affected patients. It also does not require these entities to limit the patients who receive the discounted drugs to those who are uninsured or underinsured.
As noted earlier, 340B discounts are not counted in the manufacturers’ best price, and they are also exempt from formulas that set reimbursement for fee-for-service Medicare Part D and Part B, in order to better reflect their acquisition costs. These exemptions in turn influence the costs of drug therapies among fee-for-service Medicare beneficiaries and the commercially insured. Patient deductibles and coinsurance payments associated with prescription drugs reflect the reimbursement set by the insurer to the pharmacy or the clinic; these are unaffected by 340B discounts.
Debate about the program has intensified recently, due in part to the large number and the significant diversity of providers receiving the discounts and their safety net roles (GAO, 2011; OIG, 2011; von Oehsen et al., 2012). Outpatient clinics participating in 340B are, by definition, serving vulnerable patient populations. These participants include federally qualified health centers, critical access hospitals, rural referral centers, specialized clinics (including black lung clinics, comprehensive hemophilia diagnostic treatment centers, Title X family planning clinics, sexually transmitted disease clinics, tuberculosis clinics, Native Hawaiian health centers, tribal/urban Indian health centers) and Ryan White HIV/AIDS Program grantees. In 2015, these standalone safety-net clinics were outnumbered by hospitals, their affiliated outpatient clinics, and contract pharmacies participating in 340B (OIG, 2014).
Particular scrutiny has focused on acute care nonprofit hospitals. In 2014, roughly one-third of all acute-care not-for-profit hospitals in the United States qualified as covered entities under the 340B program (Conti and Bach, 2014), and they are thought to have accounted for approximately 48 percent of the national outpatient hospital visits (Mulcahy et al., 2014). In contrast to the clinics, acute care, nonprofit hospitals and their affiliated outpatient clinics participating in the 340B program are not required to demonstrate that they provide community benefits in the outpatient setting. To be eligible for 340B discounts, HRSA requires only that hospitals provide inpatient services to Medicaid and low-income Medicare beneficiaries (to the degree that their Medicare disproportionate share patient percentage11 exceeds the eligibility threshold of 11.75 percent) (GAO,
___________________
11 Enacted by statute in 1986, Medicare’s disproportionate share adjustments were intended to provide additional reimbursement for hospitals that incur higher-than-average costs per case because they serve a significantly disproportionate share of low-income patients (CMS, 2017b). The disproportionate patient percentage is equal to the sum of the percentage of
2011). However, in the years since the program’s inception, the structure of hospitals in the United States has dramatically changed, with nonprofit hospitals increasingly displaying characteristics of for-profit hospitals (Bai and Anderson, 2016; Horwitz, 2005; IOM, 2000), and standalone hospitals pursing mergers and affiliations with other hospitals and hospital systems and outpatient provider groups (Baker et al., 2014; Cutler and Scott Morton, 2013).
Evidence about the impact of 340B revenue on safety net and community need engagement among qualifying hospitals is largely anecdotal (340B Health, 2016; Kantarjian and Chapman, 2015; Wallack and Herzog, 2011). GAO conducted a cross-sectional comparison of 340B-qualified Medicare disproportionate share hospitals with non-340B hospitals in 2012 using publicly available data from Medicare hospital cost reports (GAO, 2015). The report found that 340B hospitals provided more uncompensated care than did non-340B hospitals and also had lower profit margins than non-340B hospitals, in part because they provided more uncompensated and charity care. A more recent report found that hospitals participating in 340B in 2015 exhibited widely varying financial stability and safety net care provision (Nikpay et al., 2017). Some 340B disproportionate share hospital (DSH) program participants operated at a substantial loss, but at least one-quarter of participants operated with a comfortable margin. Many of the hospitals with the highest operating margins were also those that provided the least uncompensated care, while the hospitals that provided the most uncompensated care had the lowest operating margins. Furthermore, there was little correlation between county-level uninsured rates and the adjusted DSH patient percentage.
Finally, some 340B hospitals and clinics built large networks of contract pharmacies after HRSA released its 2010 guidance. As contract pharmacy arrangements have proliferated, especially with national chains including Walgreens, Rite Aid, CVS, and Walmart, these agreements have come under scrutiny. They are not subject to routine independent audits like other 340B program providers and manufacturers. Furthermore, contract pharmacies are not required to demonstrate that they serve vulnerable populations at all, nor are they required to show that they meet the core program objectives to qualify for discounts. In 2014, the HHS Office of Inspector General released a study regarding contract pharmacies that was conducted by interviewing a “purposeful sample” of 30 administrators and representatives of covered entities (OIG, 2014). The report noted that covered entities using contract pharmacies do not always offer the discounted 340B price
___________________
Medicare inpatient days attributable to patients eligible for both Medicare Part A and the Supplemental Security Income plus the percentage of total inpatient days attributable to patients eligible for Medicaid but not Medicare Part A.
to uninsured patients and that covered entities did not “conduct all of the oversight activities recommended by HRSA.”
Under current statute, neither HRSA nor CMS collects information from qualifying entities or drug manufacturers regarding which drugs are being purchased through the 340B program, the amount of 340B-derived revenue generated by qualifying entities, or how revenues are used to benefit vulnerable patient populations. Reports from several pharmaceutical manufacturers suggest that sizable proportions of national product sales (10 to 20 percent) are currently subject to 340B discounts. The sales of drugs through one program vendor totaled $12 billion in 2012, and $2 billion of Genentech’s sales flowed through the 340B program in 2016 (Fein, 2017). A 2011 GAO report documented that some covered entities in the 340B program generated revenue that “exceeded drug-related costs,” while others did not (GAO, 2011). GAO also analyzed spending on oncology drugs covered under Medicare Part B in 2012, comparing hospitals that were or were not qualified to participate in the 340B program (GAO, 2015). Part B spending on those drugs was substantially higher at 340B hospitals than at non-340B hospitals. These differences did not appear to be explained by the limited number of hospital characteristics examined or by patients’ health status.
Stakeholders have expressed concern that the scale of the program has increased without a subsequent correlation in resources dedicated to oversight. In the past several years, HRSA has been working to release a comprehensive update to the program’s definitions, covered entity qualifications and program participation requirements for covered entities and manufacturers, sometimes referred to as the omnibus Mega-Reg (HRSA, 2015). Of particular concern has been the strengthening of oversight by HRSA and CMS to adequately enforce existing prohibitions on diversion and duplicate discounts among covered entities and contract pharmacies (GAO, 2011). Diversion is when a 340B drug is given to an ineligible patient or resold by the covered entity. Under current statute, eligible patients are defined as those who receive regular medical care at covered entities or who participate in an AIDS drug-purchasing assistance program and who are not insured by Medicaid, although there are some exceptions. Duplicate discounts occur when a covered entity receives the 340B discount and the state receives a Medicaid drug rebate, also from the drug’s manufacturer, on the same unit. While manufacturers can audit covered entities for suspected unauthorized use of 340B drugs, covered entities do not have any audit authority and they must petition HRSA to investigate manufacturers or turn to the judicial system when purported violations in 340B pricing occur; therefore, another focus of these efforts has been to strengthen oversight of possible manufacturer overcharges. The ACA required a new dispute resolution process and greater pricing transparency by establishing a 340B
pricing database; however, while HRSA has started these initiatives, it has not finalized them. In February 2017, the Trump administration cancelled the release of the Mega rule (Ellison, 2017), effectively pushing 340B reform into the purview of CMS and the U.S. Congress. In November 2017, HHS finalized a rule that would reduce Medicare Part B’s reimbursement for hospital outpatient clinics’ use of 340B-discounted drugs and increase oversight of the program. This change will also result in reduced out-of-pocket payments for Part B beneficiaries undergoing outpatient drug-based treatment (HHS, 2017).
RARE DISEASES
The special protections afforded to drugs that prevent or treat rare diseases also influence their availability and may have an impact on their affordability as well. The Orphan Drug Act was passed in 1983 as an amendment to the Federal Food, Drug, and Cosmetic Act. A 1984 amendment to the act defined rare diseases as those affecting “less than 200,000 persons in the United States” and for which “there is no reasonable expectation that the cost of developing and making available in the United States a drug for such disease or condition will be recovered from the sale in the United States.”12 Based on the 2017 U.S. population, that translates to approximately 6.1 in 10,000 people. The European Union identifies a rare disease as a condition affecting no more than 5 in 10,000 people (Gammie et al., 2015).
Since 1983, more than 600 drugs and biological products have been brought to market with the act’s assistance (FDA, 2017e). Fewer than 10 such industry-sponsored products entered the market in the decade preceding the act (FDA, 2017e). Over the past 5 years, orphan drug approvals have increased exponentially (Evaluate Pharma, 2017). In 2016, nearly half of the new medications approved were orphan drugs, including two that are indicated for diseases with no approved treatments (FDA, 2017e).
The program provides a number of benefits to the sponsors of FDA-designated products for rare diseases (FDA, 2017e), including an additional 7 years of market exclusivity. Participating firms also benefit from more open study protocols (with fewer eligibility criteria), which are intended to increase access of affected patients to the medications, and these firms may also receive modest FDA grant support to investigate treatments for rare diseases. The regulatory review process for orphan drugs is expedited, and clinical trials can enroll smaller numbers of patients than would otherwise be acceptable in registration trials. The manufacturers of orphan drugs can
___________________
12 Health Promotion and Disease Prevention Amendments of 1984, Public Law 98-551, 98 Stat 2815 (1984), § 4.
also qualify for tax credits to help support testing. Furthermore, the act allows manufacturers to claim a tax credit13 in the taxable year of up to 50 percent for expenses paid or incurred by the sponsor on human clinical trials required to obtain FDA approval (FDA, 2017c). These factors tend to substantially reduce the development costs for orphan drugs compared with what traditional drugs cost to develop (HHS, 2016). However, sponsors are required to request orphan drug designation from the FDA before filing a new drug application (FDA, 2017e).
Drugs for rare diseases often have higher prices because of the small size of the eligible patient population and because there are generally few if any competitors to address what is most often a high unmet need. Orphan drugs are also more likely to be biologics, which tend to be less susceptible to generic competition (Thomson Reuters, 2012). The median cost per patient is 5.5 times higher for orphan drugs than for non-orphan drugs (Evaluate Pharma, 2017). In 2016, the median annual cost for an orphan drug in the United States was more than $32,000, although the 10 therapies used by the most patients averaged less, at $14,909 (QuintilesIMS, 2017). Among the top 100 drugs in the United States, the average cost per patient per year for orphan drugs was $140,443 in 2016, compared with $27,756 for a non-orphan drug (QuintilesIMS, 2017).
From 2015 to 2016, orphan drug sales increased 12.2 percent to $114 billion, compared with an increase of 2.4 percent (to $578 billion) for non-orphan drug sales (Evaluate Pharma, 2017). In 2016, of the total drug sales ($450 billion) in the United States, approximately 7.9 percent of total spending was for orphan indications of approved orphan drugs, up from 3 percent in 1993 (QuintilesIMS, 2017). By 2020, estimated worldwide sales are projected to reach $209 billion (approximately 21 percent of prescription sales excluding generics) (Evaluate Pharma, 2017).
The Orphan Drug Act has recently come under increasing scrutiny for several reasons, including (1) the fact that orphan drug status has been bestowed on some drugs that were already available (and thus did not need orphan drug program benefits to make it to market), and (2) the way that some manufacturers have selected a subset of eligible patients to qualify for orphan drug status and then extended the scope of the drug’s use to a broader population that exceeds the Orphan Drug Act limits of 200,000 potential patients—a practice termed by critics as “salami slicing” (Daniel et al., 2016; Kesselheim et al., 2017; Loughnot, 2005; Pulsinelli, 1999).
Some drugs receiving orphan drug status have in fact become “blockbuster” successes, with more than $1 billion in annual sales. These include Vioxx, Cialis, and Botox. Rituximab, the highest-selling orphan drug to
___________________
13 A bill introduced in the U.S. House of Representatives in October 2017 would eliminate this tax credit (H.R. 1, Subtitle E, section 3401).
date, is a biologic (monoclonal antibody) originally intended for treating lymphoma. It is now used to treat a wide variety of conditions, including non-Hodgkin’s lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis, and several skin disorders. In 2016, 7 of the 10 best-selling drugs in the United States had at one time received orphan drug status (Evaluate Pharma, 2017).
Some drugs have received multiple orphan designations, each of them creating a new 7-year market exclusivity. For example, Gleevec has received 9 separate orphan drug designations and had a reported $3.3 billion in sales for 2016 (Novartis, 2017). A 2017 investigation found that 70 drugs (more than 10 percent of the total approved for orphan status) had received their status after having already been approved for marketing by the FDA (Tribble and Lupkin, 2017). This group included the blockbuster drugs Crestor (for cholesterol control), Abilify (for psychiatric disorders), and Humira (for rheumatoid arthritis). The investigators found that another 80 drugs had received multiple orphan designations and that, overall, approximately one-third of all orphan drug approvals either had been for repurposed drugs already marketed or had received multiple orphan drug designations (and hence multiple program benefits). According to another recent analysis, 98 drugs with orphan status also had non-orphan indications. Of those, 54 received a non-orphan indication first and 10 received both orphan and non-orphan indications simultaneously (QuintilesIMS, 2017).
While special incentives are unquestionably needed to justify firms pursuing drugs with very small markets, empirical evidence indicates that the current orphan drug program may be misused.
OTHER INFLUENCES
Awareness of Costs
In order for clinicians and patients to make optimal choices relating to drug therapies, both must have reliable information regarding the benefits and costs of both the drug under consideration and other treatment options. However, given the large and expanding array of choices, expecting even experienced clinicians to have a full grasp of the benefits and the risks of all reasonable alternatives is at best problematic, while the patients themselves can be expected to know and understand far less than the providers.
Price-informed care has been shown to reduce unnecessary medical spending (Stammen et al., 2015). However, at present, clinicians are generally unaware of the actual cost of medications—either to health insurers or to the patients (Schutte et al., 2017). A review of studies of clinician awareness of medication costs found that clinicians were able to estimate
drug costs within 25 percent of the true cost less than one-third of the time. Clinicians tended to underestimate the cost of expensive medications and to overestimate the cost of cheaper medications (Allan et al., 2007).
Clinicians’ unfamiliarity with medication costs is not due to a lack of interest or concern. The vast majority of prescribers report that it is important to manage the patient’s out-of-pocket medication costs (Shrank et al., 2006a). But the challenge is not a simple one: developing an awareness of prices is complicated by the existence of multiple formularies, differences in list prices, and differences in individual patients’ insurance plans. A decade ago, prescribers reported that they were already facing challenges dealing with the overwhelming quantity of medical information (Shrank et al., 2006a), and the situation has likely intensified. Hence, clinicians are unlikely to respond favorably to anything that requires them to deal with multiple prices for each patient’s medication unless the information is provided in an efficient, user-friendly fashion at the point of care.
The use of high-cost medications can be reduced if prescribers have ready electronic access to information relating to the medications that are prompted by the diagnosis, in addition to relevant summaries of product safety and cost pertaining to the insurance plan (McMullin et al., 2005). An increasing number of firms are creating Web- and smartphone-based applications that can be used by prescribers and patients to view drug cost information based on list prices. However, these list prices generally reflect what the patient would pay without insurance and may be very different from what the actual out-of-pocket payments would be according to the patient’s insurance plan.
Patients themselves are often unaware of the amount that they will pay for their medications. The “sticker shock” at the pharmacy leads to lower fill rates for prescriptions, and some patients extend their medications by reducing dosages. Some health plans and PBMs are attempting to improve patients’ access to medication cost information. Insurance companies are creating Web portals designed to help patients learn about which medications will be covered under their plans with a lower copayment, when cheaper alternatives exist, and whether patient assistance programs are available (Humana, 2017). A complicating factor for both patients and the insurers is that insurers typically do not have access to information about rebate and discount agreements between manufacturers and PBMs.
Also, more prescribers are requesting that pharmacists routinely be involved in the process of reviewing the costs of medications (Ross, 2016). Indeed, the Medicare Modernization Act established the requirements that Part D sponsors14 must offer each enrolled beneficiary a minimum level of
___________________
14 CMS contracts with sponsors (insurers) to provide the Medicare Part D prescription drug benefit.
medication therapy management services that includes interventions for both the Medicare beneficiaries and prescribers. The sponsors must also offer at least once per year an interactive person-to-person comprehensive medication review by a pharmacist or other qualified provider. They must also perform quarterly medication reviews with follow-up interventions when necessary, focusing on patients with high expected out-of-pocket costs who have multiple chronic conditions and who are taking multiple medications. These strategies have been linked to reductions in medical costs (Ramalho de Oliveira et al., 2010) and improvements in medication adherence (Pringle et al., 2014).
State Laws on Prescription Form Language
State laws that specify the language used in prescription forms can also influence prescriber behavior. Even in situations when a generic medication is available, prescribers can mandate that a brand medication is dispensed by indicating “dispense as written” on the prescription. However, due to the increased cost of brand medications, the use of “dispense as written” has been shown to reduce the likelihood that the patient will actually purchase the medication and, consequently, take the needed medication. A pharmacy claims analysis found that patients with a tiered pharmacy benefit who received a generic medication were 62 percent more likely to use their medications appropriately than those who had been prescribed more expensive medications (Shrank et al., 2006b). Another analysis estimated that if the 5 percent usage of “dispense as written” observed in the study sample was extrapolated to the United States as a whole, the result would be an additional $1.2 billion in out-of-pocket spending by patients and an additional $7.7 billion in drug costs for health systems (Shrank et al., 2011).
The design of the prescription pad itself can influence the likelihood that a prescriber uses the “dispense as written” designation. If the provider must check a box in order to designate “generic substitution is permitted,” more prescriptions are filled with branded medications. Conversely, if the prescriber must check a box saying “dispense as written,” then more prescriptions are filled with generic drugs (Helmons et al., 2014). This result is consistent with the literature on behavioral sciences that shows how answers to a question can be “framed” by the manner in which the question is presented. Some approaches to reducing the use of “dispense as written” designations that are being tested include improving the education of providers and patients on the clinical equivalence of generic medications, and imposing financial penalties on clinicians by health plans (Shrank et al., 2011).
Patients may themselves request that a brand medication be dispensed rather than the generic equivalent when they reach the pharmacy. An
analysis of state Medicaid programs with “mandatory” generic substitution programs compared states that required patient consent for generic substation versus those that did not require patient consent. The study found an increased use of branded medications in states that required patient consent. States that required patient consent paid on average an additional $15 per prescription (Shrank et al., 2011).
Payments to Prescribers
The norms of medical professionalism obligate clinicians to make prescribing decisions that are in their patients’ best interests. Payments to clinicians by pharmaceutical manufacturers, which include speaking honoraria, travel expenses, and paid meals are seen by some as creating a conflict of interest. These payments could potentially lead clinicians to favor medications from a sponsor over non-sponsored medications, even when the sponsored medications are less effective or more expensive. A 2009 Institute of Medicine report concluded that industry payments to clinicians were likely to create conflicts of interest that were not outweighed by the positive benefits of working with drug makers, such as continuing education (IOM, 2009). The report recommended that clinicians not accept industry payments, and that industry (including pharmaceutical companies) refrain from offering payments to clinicians. This report also recommended public reporting and disclosure of potential conflicts of interest although this is not a sufficient remedy by itself.
The Open Payments program (also known as the Physician Payments Sunshine Act) within the ACA requires industry to disclose payments made to clinicians in a public database. The Physicians Payments Sunshine Act has increased the visibility of the prevalence of industry payments to clinicians and the association between industry payments and prescribing behavior. A recent study of clinicians in Massachusetts, using data from the Sunshine program on payments, found an association between payments by statin manufacturers to clinicians and an increased likelihood of prescribing branded statins. Furthermore, payments for the educational training of clinicians were associated with a 4.8 percent increase in the rate of brand-name prescribing (Yeh et al., 2016). Another study found that having received an industry-sponsored meal was associated with higher likelihood of prescribing branded pharmaceuticals across a range of therapeutic classes (DeJong et al., 2016). Findings from these studies show a potential influence of such benefits on prescribing choices.
In principle, disclosure has the potential to reduce conflicts of interest by creating greater scrutiny of financial relationships between clinicians and industry. However, it seems likely that most patients have limited knowledge about transparency tools (including the Open Payments database), so
the use of these tools by patients is likely to be limited (Ross, 2017). And, contrary to what one might expect, the disclosure of conflicts of interests can increase the amount of biased recommendations offered by clinicians and reduce skepticism from patients, who may believe that disclosure increases trustworthiness (Loewenstein et al., 2012). In focus groups about the open payments system, clinicians have expressed general appreciation for such transparency (Chimonas et al., 2017).
Steps to further limit pharmaceutical industry influence on clinicians have been limited. Some health systems have rules that limit interactions between drug company representatives and health system employees. These include anti-detailing policies, which prohibit direct marketing to these employees. One study found that the adoption of these policies in an academic medical center reduced the controversial practice of providing off-label prescriptions (uses that have not been approved by the FDA) of antipsychotic medications to children (Larkin et al., 2014). Some states had adopted laws, including transparency laws, that preceded the federal open payments legislation and bans on clinicians accepting gifts. One study found that the market share of new, costly drugs was substantially lower in such states than in states without the laws (King and Bearman, 2017). While legal concerns have been raised about the commercial speech rights of drug manufacturers to promote their products, a key legal issue is whether laws that restrict some corporate speech act to advance the public’s interest (Kesselheim and Avorn, 2008).
Prescriber Reimbursement
Another potential conflict of interest arises from the current percentage-based reimbursement system for drugs administered in outpatient clinics, under which the use of higher-priced drugs results in higher payments to providers. Specifically, under the buy and bill arrangement in which clinicians are reimbursed for the average sales price of the drug plus 6 percent plus an administration fee, the spread between the payer-reimbursed price and the acquisition cost of the drug generates revenue for standalone and hospital-affiliated clinics (Conti et al., 2013; Howard et al., 2015; Malin, et al., 2013; Polite et al., 2014; Shahinian et al., 2010). In oncology, where the infused or injected administration of drugs in the outpatient setting is very common, a substantial fraction of practice revenues may depend on the use of these drugs.
As a consequence, studies suggest that oncologists’ drug choices are responsive to drug-based profit margins (Conti et al., 2012; Jacobson, 2006; Jacobson et al., 2010). When Medicare reimbursements for these drugs were reduced under the Medicare Modernization Act (MMA), the volume of chemotherapy use increased—which suggests that the total dol-
lars received remained the same for prescribing doctors (Jacobson et al., 2010). Furthermore, eligibility for special discounts on the acquisition costs of these drugs that do not affect payer reimbursement for their use may act to alter the incentives for community providers to remain independent. For example, hospital-affiliated outpatient practices that qualify for 340B discounts can purchase drugs at reduced cost while still receiving full reimbursement for them in addition to their ability to charge facility fees. Conversely, community oncology practices that do not qualify for 340B discounts operate on lower per person-per treatment margins derived from the administration of the drugs they purchase, including the revenue generated from buy-and-bill reimbursements and the ability to charge facility fees (Polite et al., 2014). These disparities in revenue-generating incentives may act to encourage the consolidation of health care providers (Baker et al., 2014; Cutler and Scott-Morgan, 2013). For example, there has been significant growth in 340B eligibility among outpatient clinics affiliated with 340B-participating hospitals preceding and following ACA implementation. As a result, GAO estimates that 340B discounts apply to 50 percent of cancer drugs sold and paid for by Medicare Part B (GAO, 2015). For drugs dispensed or used by clinicians at a hospital-affiliated clinic or an outpatient infusion center affiliated with a hospital, these providers also charge payers facility fees, which may amount to 50 percent or more of the drug’s acquisition cost. As the site of care for outpatient infusion services has increasingly shifted toward hospital-owned or affiliated practices in recent years, spending associated with this form of care has grown (MedPAC, 2017b).
Unlike drugs covered under insurers’ pharmacy benefits, the coverage of drugs under the medical benefit is essentially guaranteed for indications approved by the FDA and for many off-label uses as well (Bach, 2009; Conti et al., 2013; Scheingold et al., 2017). Formularies and other supply-side coverage restrictions based on evidence of clinical benefit or cost-effectiveness are not commonly used to restrict wasteful spending on these drugs. Many state laws, affecting about three-quarters of the U.S. population, require insurance coverage of infused and injected cancer treatments if their use is recognized in drug compendia, the peer-reviewed literature, or both (Bach, 2009; IOM, 2013). However, the quality of information in compendia is often variable and adequate evidence is often lacking (Abernethy et al., 2010). This complex legal and regulatory framework makes it difficult for payers to use comparative effectiveness evaluations in reimbursement decisions for cancer drugs (Pearson, 2012). Thus, under buy and bill, medical providers face incentives to use expensive prescription drugs, often in combination, whenever indicated (Howard et al., 2015). This system also creates a disincentive to substitute lower-priced drugs that offer patients equivalent outcomes or to substitute generics for more costly branded drugs (Conti et al., 2012).
Buy and bill also creates incentives for high pricing of drugs covered under the Part B benefit. As noted by Brock (2010), manufacturers know that expensive new cancer drugs will not be denied coverage by payers on the basis of cost, so they have no incentive to set prices to meet any cost-effectiveness standard. CMS posts a new average sales price every quarter based on information submitted by drug manufacturers 6 months earlier. As a result, clinician reimbursement remains stagnant for two quarters after drug acquisition costs rise, posing a financial risk for outpatient practices (Conti et al., 2013). Howard and colleagues (2015) argued that the launch prices of these drugs are high and have grown over time in part because manufacturers understand the risks practices face if prices rise after launch.
Over the years, there have been many proposals to minimize the influence of drug-derived revenue on clinician prescribing behavior in the Medicare program (Bach, 2009; Polite et al., 2014). This was one motivating rationale behind the MMA’s revision of Medicare Part B payment to average sales price—this policy explicitly linked reimbursement to the drugs’ actual acquisition cost, including the availability of volume-based discounts and rebates. Proposed alternative methods for setting alternative reimbursements under Medicare Part B have included invoice pricing, least costly alternative reimbursement, the bundling of drugs into episode-of-care payments, shifting Part B drugs to the Medicare Part D benefit, and the revision of the failed Competitive Acquisition Program enacted under MMA (Polite et al., 2014, 2015). Under the Bipartisan Budget Act of 2015, CMS will implement Section 603, which specifies that services provided at off-campus hospital outpatient departments that began billing under the Medicare outpatient prospective payment system on or after November 2, 2015 will no longer be reimbursed under outpatient rates (CMS, 2016). This site-neutral payment policy was designed to reduce Medicare spending on off-campus hospital outpatient department services that could be performed at a physician’s office for a lower rate. These changes will be phased in over 4 years beginning in 2017. Payments for services provided at off-campus hospital departments that began billing Medicare before this date are not covered under this policy (OIG, 2014).
In 2016, the CMS Innovation Center created new authority for payment demonstration projects and unveiled plans for the Medicare Part B Drug Payment Model. This demonstration was intended to test in two phases the effect of alternative payment models on Part B spending across therapeutic categories. In the first phase, an alternative to the 6 percent markup would be tested by including a lower-percentage markup, offset by a flat daily supplemental payment of $16.50. In the second phase, payment alternatives such as reference pricing (pegging reimbursement to the least expensive available drug in the class) and outcome-based risk sharing (pro-
viding higher reimbursement for more favorable patient outcomes) would be introduced. The CMS demonstration was designed to be cost-neutral in the short run, but ultimately to identify models that would produce greater system cost savings (Schrag, 2016). The Part B demonstration was canceled before implementation began (Dolan, 2016). However, there is a relevant demonstration project under way in the Oncology Care Model, wherein clinician practices have entered into payment arrangements that include financial and performance accountability for episodes of patient care surrounding chemotherapy administration.
FINDINGS
Based on the material presented in this chapter, the following findings are offered:
Finding 3-1: Position statements provided by various participants in the drug pricing debate reveal numerous instances of potential conflicts of interests, including various combinations of financial relationships among biopharmaceutical companies, patient advocates, academic researchers, health care professionals, and their representative organizations.
Finding 3-2: Publicly available evidence shows that the biopharmaceutical industry has higher profitability than other comparable sectors in the economy.
Finding 3-3: There is a widespread disagreement about the actual costs underlying biopharmaceutical research and development and the proper methods to calculate them.
Finding 3-4: When branded drugs go off patent and a generic supplier enters the market, prices for those medications usually decline; with two or more generic suppliers, the market prices generally decline significantly.
Finding 3-5: Mergers between companies that produce both branded and generic drugs treating the same condition can, in absence of other competition, have anti-competitive effects that often result in undesirable price increases.
Finding 3-6: Delays and backlogs in the U.S. Food and Drug Administration approval of generics and biosimilars curtail market competition and thereby increase the likelihood of higher drug prices.
Finding 3-7: In the absence of evidence of harm (as opposed to the concerns surrounding potential harm) with respect to importation of generics and biosimilars when competition is lacking, and given the potential cost-savings for patients, policy experiments related to generic and biosimilar importation could be useful.
Finding 3-8: Essential medicines lists by other OECD countries have been generally helpful in managing the availability and affordability of drug therapies.
Finding 3-9: Drug shortages occur regularly and can lead to adverse outcomes for patients.
Finding 3-10: The list prices provided by manufacturers can significantly affect patients’ drug costs and access. This affects retail purchases of both insured and uninsured patients, as well as drugs purchased by clinicians and hospitals and administered to patients directly.
Finding 3-11: Current insurance benefit designs for prescription drugs often expose consumers to considerable financial risk and can unfavorably affect patients’ medication adherence.
Finding 3-12: Large biopharmaceutical companies spend substantially more on marketing and administration than on research and development that could lead to new drugs.
Finding 3-13: Direct-to-consumer advertising of prescription drugs has increased substantially over time and can adversely influence consumer choices.
Finding 3-14: While copay coupons provided by pharmaceutical companies can expand patient access to high-cost medications, they also increase the percentage of prescriptions that are filled with branded drugs, increase overall drug spending, and drive up individuals’ insurance premiums.
Finding 3-15: Programs promulgated under the Orphan Drug Act—which were originally designed to foster the development of innovative drugs for rare conditions—have expanded well beyond their original intent and are counteracting efforts to make medicines more affordable.
Finding 3-16: Section 340B of the U.S. Public Health Service Act had the stated intent of improving the access of low-income populations
to medicines at discounted rates; however, it is unclear whether the benefits of the program flow to the intended vulnerable populations. As implemented, the program has expanded well beyond assisting low-income patients and may therefore be acting to increase the cost of medicines paid by insurers and patients.
Finding 3-17: Current insurer reimbursement policies for clinician-administered drugs in the outpatient setting minimize incentives for medical providers to select treatments and settings for patient care that are the most cost-effective. These policies may serve to inflate the prices of these drugs charged by manufacturers and other members of the supply chain who profit from the current system, and put patients at clinical and financial risk.
Finding 3-18: In order for both consumers and clinicians to make well-informed decisions regarding prescription drug therapies, reliable and objective information is needed—including information on potential clinical outcomes, the comparable effectiveness of alternative treatments, and out-of-pocket and overall costs to the patients. Some but not all of this information is available today; particularly lacking is information regarding costs.




















































