
10
Cancers
In prior Surgeon General reports, active smoking of combustible tobacco cigarettes has been determined to be causally associated with increased risk of 13 different malignancies (HHS, 2014). The biological mechanisms driving combustible tobacco cigarette smoking as a cause of such a diverse spectrum of cancers is due in large part to the wide array of carcinogens present in combustible tobacco cigarette smoke, many of which are generated by the combustion of the tobacco. There are more than 7,000 chemicals in combustible tobacco cigarette smoke, and more than 70 are established human carcinogens (HHS, 2010; IARC, 2012). In addition to combustible tobacco cigarette smoking, pipe and cigar smoking are established causes of lung cancer (HHS, 2014). Furthermore, even exposure to secondhand tobacco smoke, which results in much lower levels of smoke exposure than active smoking, is causally associated with lung cancer (HHS, 2006).
The cancer risk associated with the use of electronic cigarettes hypothetically would be expected to be less than combustible tobacco cigarettes based on the rationale that e-cigarettes include nicotine from tobacco—but not all the other tobacco constituents—and would therefore result in a reduced burden of carcinogens delivered to the user. Additionally, the nicotine present in e-cigarette aerosols does not contain appreciable amounts of tobacco-specific nitrosamines, nor are other pyrolysis products from nicotine formed. Moreover, compared with combustible tobacco smoke, potentially carcinogenic components of e-cigarette aerosols may be orders of magnitude less “carcinogenic” compared with those present
in tobacco smoke (Chen et al., 2017; Stephens, 2018). (Comparisons of combustible tobacco smoke and e-cigarette aerosols are described in more detail in Chapter 18 on Harm Reduction.) By contrast, there is uncertainty about the potential mutagenicity and carcinogenicity of other e-cigarette substances, such as flavorants and humectants, present in the aerosol emitted from e-cigarettes that result from the heating and aerosolization of the liquid in these products. Furthermore, as described in Chapter 5, carcinogens such as formaldehyde and arsenic have been detected in electronic cigarette aerosol.
CHARACTERIZATION OF DISEASE ENDPOINTS AND INTERMEDIATE OUTCOMES
As shown in Figure 10-1, the etiology of cancers induced by environmental exposures is a complex, multistep process that generally takes years to develop. There are several biologically plausible pathways for
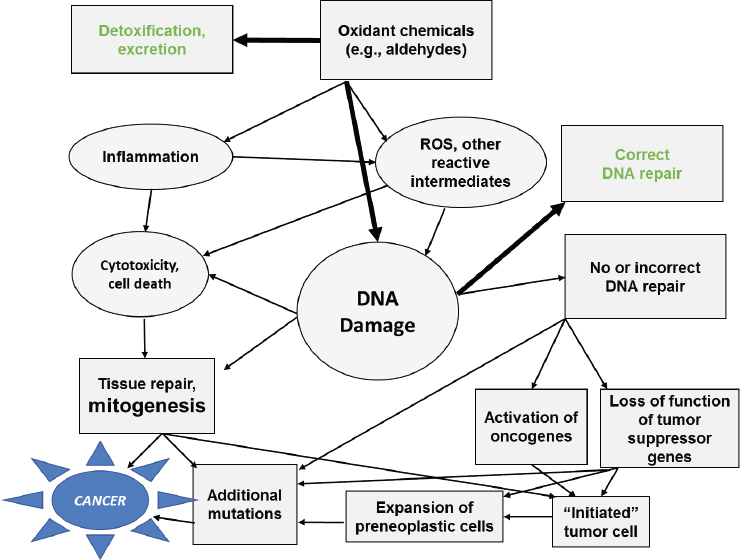
NOTE: ROS = reactive oxygen species.
which components of e-cigarette aerosols could conceptually influence cancer development. As discussed in Chapter 5, numerous compounds identified in e-cigarette aerosols can form reactive oxygen species (ROS), and/or can be converted to reactive intermediates capable of binding to DNA. Oxidative damage to DNA, and/or the direct adduction of reactive molecules to DNA, such as can occur with formaldehyde, is the most important intermediate outcome of chemical carcinogenesis. As discussed later in this chapter, some studies have identified cytotoxicity of e-cigarette aerosols, potentially contributing to tissue repair and mitogenic response, which is another important pathway in the development of chemically induced cancers. Formaldehyde is perhaps the most prevalent component of e-cigarette aerosols capable of inducing ROS formation. A 2014 National Research Council report on the carcinogenesis of formaldehyde determined that epidemiological evidence was strongest for an association between formaldehyde exposure and cancers of the nasopharyngeal region and sinonasal cavities and myeloid leukemia (NRC, 2014). However, it should be recognized that formaldehyde is highly reactive, and potential DNA damage induced by it is most likely to occur at the site of exposure, the upper airways. It is also formed in small amounts by endogenous processes, so whether toxicologically significant amounts of formaldehyde from exogenous exposures can cause DNA damage in tissues distant from the site of exposure is controversial (Swenberg et al., 2011). Nevertheless, the presence of levels of formaldehyde in e-cigarette aerosols at concentrations that reportedly can exceed occupational exposure limits by an order of magnitude or more (see Chapter 5) are of concern for the potential risk to nasopharyngeal and lung cancer. It should be noted that, as described in earlier chapters, the levels of formaldehyde in e-cigarette aerosols can vary by many orders of magnitude, depending in large part on the device parameters (e.g., power), e-liquid contents (e.g., propylene glycol [PG] and glycerol), and user characteristics (e.g., puff topography). Thus, the presence of formaldehyde and other potentially mutagenic and cytotoxic constituents provides biologically relevant mechanisms whereby long-term use of e-cigarettes could affect cancer risk.
OPTIMAL STUDY DESIGN
The strongest evidence to characterize the potential association between e-cigarette use and the risk of human cancer will be methodologically rigorous epidemiological studies with human cancer as the outcome. Importantly, many e-cigarette users will be current or former combustible tobacco cigarette smokers, especially in the near term, and the effects of current and former smoking will be a challenging confounder to account
for in observational studies. The second strongest level of evidence will be studies of intermediate cancer endpoints; for example, a study of e-cigarette use in relation to colorectal adenomas would have direct relevance to colorectal cancer because adenomas are precursor lesions in the colon carcinogenesis pathway. An important result of this comprehensive review is that the published literature is currently devoid of any evidence that includes rigorously designed epidemiological studies that include intermediate cancer endpoints, let alone cancer as an endpoint. Except for a study that included self-reported cancer as an adverse event (Manzoli et al., 2017), the published data have used biomarkers (oxidative stress and inflammation) as study outcomes.
EPIDEMIOLOGY
The literature search identified two studies on e-cigarette products in humans that refer to cancer. One of these was the study by Manzoli and colleagues (2017), which was composed of three groups (total n = 932) with the following sample sizes at the end of the 24-month follow-up: smokers of only combustible tobacco cigarettes throughout follow-up (n = 363), users of only e-cigarettes throughout follow-up (n = 97), and users of both e-cigarettes and combustible tobacco cigarettes throughout follow-up (n = 37). The authors defined e-cigarette users as users of any type of e-cigarette for 6 months or more. The authors report “any cancer” under serious adverse events, with the following results: only combustible tobacco cigarettes 0.8 percent (3/363), only e-cigarettes 2.1 percent (2/97), and dual users, 0 percent (0/37). The risk ratios the committee calculated from these data, using combustible tobacco cigarettes only as the referent category, are 2.49 (95% CI = 0.42–14.72) for e-cigarettes only and 0 (95% CI not estimable) for dual use. The results do not provide any indication for cancer risk reduction from sole use of e-cigarettes. These data are extremely limited by sample size and are of low quality; for example, the cancer data are presumably self-reported and not pathologically confirmed, the sample size is very small to assess the endpoint of any cancer and precludes assessment of specific malignancies, and there is no consideration of complete combustible tobacco cigarette smoking history or potential confounding factors.
In another human study, oral cells were collected by scraping the oral mucosa; the micronucleus assay was then applied to these oral mucosa cells (Franco et al., 2016) as a biomarker of potential genotoxicity and/or chromosomal instability (Luzhna et al., 2013). The Franco and colleagues (2016) study population had a total of 65 participants from three groups: (1) combustible tobacco cigarette smokers (n = 23); (2) e-cigarette users (defined as use of any e-cigarette device and liquid; n = 22); and
(3) non-smokers of combustible tobacco cigarettes and e-cigarettes (n = 20). The results revealed that compared with non-users of e-cigarettes and non-smokers of combustible tobacco cigarettes, the mean number of micronucleated cells/1,000 cells was 21 percent higher in e-cigarette users and 160 percent higher in combustible tobacco cigarette smokers. The results were also presented for the measure of total micronuclei/1,000 cells; compared with non-smokers of combustible tobacco cigarettes and e-cigarettes, e-cigarette users had mean levels that were 133 percent higher and combustible tobacco cigarette smokers had mean levels that were 633 percent greater. The pattern of associations for both micronuclei measures presented were thus consistent in showing that the average micronuclei burden was elevated in e-cigarette users relative to that in never smokers, and was elevated fourfold or more in combustible tobacco cigarette smokers compared with e-cigarette users. The only p-values reported were for the comparison of combustible tobacco cigarette smokers with e-cigarette users; these differences were statistically significant for both mean micronucleated cells/1,000 cells (p = 0.001) and total micronuclei/1,000 cells (p = 0.004). Weaknesses of this study include not presenting any evidence on the reliability of the micronucleus assay, not presenting all relevant p-values, and the lack of consideration of potential confounding factors even though the e-cigarette user group was on average 10 years older than the other study groups. This latter point is important because other studies have demonstrated age-related associations with micronuclei formation (Bonassi et al., 2011; Fenech et al., 2011).
Also relevant to a consideration of the potential association between electronic cigarettes and cancer are studies of e-cigarette use in relation to oxidative stress and inflammation; both of these biomarkers have been reviewed in detail earlier in this report. For the study of oxidative stress, the detailed results were not presented, but the graphical evidence presented failed to show a clear association between active and passive combustible tobacco smoking or active and passive e-cigarette use on acute measures of oxidative stress in an experimental setting (Poulianiti et al., 2016). A major limitation of the study by Poulianiti and colleagues is that the well-established role of combustible tobacco cigarette smoking in increasing oxidative stress (HHS, 2004, 2010) was not observed, raising major questions about the study’s validity. Whether this was due to problems with the research protocol or suboptimal assay quality is unclear.
The study of inflammation (Flouris et al., 2012) was embedded within the exact same study as the aforementioned study of oxidative stress (Poulianiti et al., 2016), in that it was the identical study population except the assay results were presented for markers of inflammation (i.e., same study, two different publications). Once again, the complete results were not presented, but the authors reported that under the experimental con-
ditions, both active tobacco smoking and secondhand exposure to tobacco smoke were significantly associated with increased circulating concentrations of inflammatory markers, including leucocytes, lymphocytes, and granulocytes (Flouris et al., 2012). These associations are consistent with the known effects of tobacco smoke exposure (HHS, 2004, 2010), and thus these findings suggest greater internal validity than for the oxidative stress results from this exact same study. The results did not show similar associations with e-cigarettes, which were not associated with these inflammatory markers.
CASE REPORTS AND OTHER CLINICAL STUDIES
Two case reports were identified that provided evidence relevant to the association between e-cigarette use and cancer. One case reported on a white male combustible tobacco cigarette smoker who had chronically elevated leucocyte and neutrophil counts in the absence of overt clinical disease; this is a clinical scenario consistent with chronic idiopathic neutrophilia (Farsalinos and Romagna, 2013). The patient was followed clinically for 6.5 years, during which he was unable to quit smoking and the symptoms persisted. The patient was then able to successfully quit smoking by using e-cigarettes. Even though the patient still used e-cigarettes after he stopped smoking combustible tobacco cigarettes, 6 months after quitting the latter, all the patient’s markers of inflammation were significantly reduced, including leucocytes, lymphocytes, neutrophils, and C-reactive protein (Farsalinos and Romagna, 2013). Case reports provide only a weak form of evidence, but this single patient’s experience suggests that e-cigarettes have less detrimental impacts than combustible tobacco cigarettes on inflammation and immune status.
In a case report published by Madsen and colleagues (2016), a 45-year-old female who used e-cigarettes presented after experiencing abdominal pain and fever for 4 months. Radiographic images revealed numerous pulmonary nodules and liver lesions consistent with extensive metastasis, but after a complete clinical workup, no evidence of malignancy was detected. A lung biopsy found an area with multinucleated giant cells. The authors reported that the biopsied lesion was consistent with a foreign-body reaction to lipophilic material. The patient subsequently stopped use of e-cigarettes; shortly thereafter, the lung nodules and liver lesions disappeared. The authors noted that the presence of multinucleated giant cells was consistent with the presence of glycerol-based oils detected in e-cigarette aerosol, and concluded that using e-cigarettes was associated with an inflammatory reaction that produces symptoms that can create the appearance of metastatic cancer.
These two case reports both relate to inflammation/immune sta-
tus, with the case report of Farsalinos and Romagna (2013) suggesting that e-cigarettes are associated with substantially less inflammation than combustible tobacco cigarettes. However, the case report of Madsen and colleagues (2016) indicated that e-cigarettes are a strong enough source of inflammation to elicit symptoms that could be misdiagnosed as a form of cancer. These case reports raise interesting questions and reinforce the long-term need for carefully designed epidemiological studies of e-cigarette use in relation to cancer risk that include appropriate comparisons based on jointly considering the use of combustible tobacco cigarettes and e-cigarettes.
IN VIVO ANIMAL STUDIES
The literature search identified no in vivo animal studies focused on the potential carcinogenic actions of long-term e-cigarette use (Dodmane et al., 2014; Haussmann and Fariss, 2016; Toth, 1982; Waldum et al., 1996).
In Vitro Mutagenicity by the Ames Salmonella Reverse Mutation Assay
Three studies used the Ames mutagenicity assay with and without S9 metabolic activation (Canistro et al., 2017; Misra et al., 2014; Thorne et al., 2016) (see Table 10-1). Canistro and colleagues (2017) evaluated urine from male rats exposed in vivo to e-cigarette aerosols. They used two different Salmonella strains: TA100, which detects predominantly base substitution mutations, and YG1024, which detects primarily frame-shift mutations. They found that urine from e-cigarette aerosol-exposed rats was directly mutagenic in TA100, and metabolic activation decreased mutagenicity in this strain. Conversely, they found that urine was not directly mutagenic in YG1024 strain, but addition of the metabolic activation system significantly increased mutagenicity of urine above the background rate in non-exposed rats. Thorne and colleagues (2016) directly tested e-cigarette aerosol collected matter (ACM) from a Vype® ePen e-liquid cartridge containing blended tobacco flavor in TA98 and TA100 strains, with and without metabolic activation at nine different concentrations, up to 2,400 µg/plate. They reported no significant increases in mutagenicity in any of the assays. Misra and colleagues (2014) also assessed two e-liquids for mutagenicity in Ames strains TA98 and TA100, with and without metabolic activation. While extracts from tobacco smoke from standard reference combustible tobacco cigarettes were mutagenic in both strains at higher concentrations (in the presence of S9), there was no increase in mutagenicity in either strain exposed to the e-cigarette aerosol extract at any dose. Thus, of three studies examining mutagenicity of e-liquids, the
TABLE 10-1 In Vitro Mutagenicity/DNA Damage Assessment of E-Cigarette Liquids and Aerosols
| Reference | Test Agents | Cells or Tissue Types |
|---|---|---|
| Breheny et al., 2017 | Vype ePen e-liquid cartridges (blended tobacco flavor) containing 18 mg/ml nicotine; comparison with tobacco smoke total particulate matter (TPM) from reference combustible tobacco cigarette (3R4F). | Bhas 42 mouse fibroblast cells. |
| Canistro et al., 2017 | e-cigarette BandZ S.r.l., (Pisa, Italy). “Essential cloud, red fruit flavor,” 20-ml package. Composition per 100 g of product: propylene glycol, glycerol, nicotine (18 mg/ml). Power set at 5.5 V, 15 W. |
Blood and urine collected from in vivo animal exposures; male S-D rats exposed via inhalation chamber. Peripheral blood for alkaline comet assay and micronucleus test. DNA extracted from lung. Urine for Ames assay, Salmonella strains TA100 and YG1024 with and without S9. |
| Misra et al., 2014 | blu e-cigarettes containing glycerol-based e-liquids, with and without nicotine and two market leader flavors (classic tobacco and magnificent menthol), were used. Combustible tobacco cigarettes (Kentucky reference 3R4F, 1R5F, and Marlboro Gold), were used for comparison. | Salmonella strains TA98 and TA100. CHO-K1 cells. |
| Dose and Time Course | Assay Employed | Results |
|---|---|---|
| Aerosol collected material (ACM) from the e-cigarette was produced using a Borgwaldt LM20X linear smoking machine with 3-second duration using a square-wave puff profile, 55-ml puff volume, 30-second frequency, 3-second puff duration. ACM concentrations 3, 6, 12, 24, 48, 60, and 120 μg/ml. Comparison to same concentrations of TPM. |
“Promoter activity” via cell transformation assay. | ACM from the e-cigarette was shown to be negative in all three promoter experiments, whereas TPM was positive in all three experiments. |
| Equivalent to 1 ml/day e-liquid. One cycle of treatment consisted of 17-second puff (6 seconds on, 5 seconds off, 6 seconds on) followed by 20-minute stop. At the end of the cycle, the animals were transferred to a clean chamber to begin the next cycle. Animals were submitted to 11 cycles per day for 5 consecutive days per week, and for 4 consecutive weeks. | Alkaline comet assay on blood. Micronucleus test on smears of peripheral blood. 8-OHdG (oxidative damage to DNA). Ames test on urine extracts. |
“Extensive DNA damage in leukocytes measured as tail comet length of the fragmented DNA determined by single- and double-strand breaks.” “Increase in the percentage of immature micronucleated reticulocytes (MN-RET) over normal reticulocyte RT.” “8-OHdG markedly increased in the lungs.” “Urine of e-cig-exposed animals induced a dose-dependent increase in the number of S. typhimurium revertants in different strains. The highest sensitivity was shown by the TA100 strain.” |
| Cells were treated for approximately 24 hours with increasing levels of e-liquids. The cellular treatment dose range used for e-cigarettes (e-liquids and pad-collected aerosols) was 0–20 mg/ml and for combustible tobacco cigarettes, 0–0.5 mg/ml. | Ames assay TA98 and TA100 with S9 activation. Micronucleus assay. | No significant induction in the number of revertants over respective controls was observed for all e-liquids. No significant induction in the MN formation over respective controls was observed for all e-liquids. |
| Reference | Test Agents | Cells or Tissue Types |
|---|---|---|
| Thorne et al., 2016 | E-cigarette ACM from Vype® ePen e-liquid cartridges (blended tobacco flavor) contained 18 mg/ml nicotine. | Salmonella strains TA98 and TA100. |
| Thorne et al., 2017 | Emissions of three aerosol products. Kentucky reference combustible tobacco cigarettes (3R4F) and 2 e-cigarette formats: (1) a puff-activated closed “cigalike” device (eStick); (2) a “closed modular” system, dual-voltage, button-activated product (ePen). | Human bronchial epithelial cells (BEAS-2Bs). |
| Welz et al., 2016 | E-liquids with the fruit flavors apple and cherry and one tobacco-flavored liquid; base mixture of 80% propylene glycol, 10% glycerol, and 10% water. All liquids had a nicotine concentration of 12 mg/ml. | Fresh tissue samples of healthy human oropharyngeal mucosa assembled into mucosal tissue cultures (spheroidal in vitro model). |
| Yu et al., 2016 | V2 e-cigarette in red American tobacco flavor and VaporFi e-cigarette in classic tobacco flavor; both brands used a mixture of 70% propylene glycol/30% glycerol liquid formulas. Both 1.2% (12 mg/ml) nicotine e-liquid and nicotine-free versions in the same flavors were used for each. | Immortalized human keratinocytes (HaCaT). HNSCC cell lines UMSCC10B and HN30. |
| Dose and Time Course | Assay Employed | Results |
|---|---|---|
| All TPM/ACM experiments were conducted using final concentrations of 0, 50, 100, 150, 200, 250, 300, 500, 1,000 and 2,400 μg/plate. | Ames assay TA98 and TA100 with S9 activation. | Non-mutagenic in the 85-mm plate incorporation assay. |
|
Average Delivered Deposition (μg/cm2).
3R4F 0 eStick 0 ePen 0 |
DNA double strand breaks (a-H2Ax immunofluorescence). | eStick and ePen were non-genotoxic and non-cytotoxic. Combustible tobacco cigarette smoke aerosols were genotoxic at a 3.1-μg/cm2 dose and cytotoxic at 26.9 μg/cm2. |
| Two different types of incubation: (1) one-time incubation for 24 hours and (2) incubation for 2.5 hours on five sequential days. DNA-damage experiments used one dose, a 15% solution of each e-liquid. | Alkaline elution DNA damage assay (neutral comet assay) | Tobacco liquid and base liquids were negative when treated once for 24 hours or when treated for 5 days. By contrast, apple and cherry liquids induced significant DNA damage in both single and repetitive 5-day treatments. |
| Single concentration 1% aerosol by volume. HaCaT cells treated for 8 weeks. UMSCC10B and HN30 each treated for 1 week. |
Neutral comet assay DNA double strand breaks (γ-H2Ax immunofluorescence). | All four e-cigarette extracts (both nicotine and non-nicotine) were positive, in all three cell types; nicotine response greater. |
two that looked directly at the e-liquid extracts did not find any evidence of mutagenicity, although the study that exposed animals (rats) in vivo to e-cigarette aerosols did find an increase in the mutagenicity of urine.
Micronucleus Assay
Both Canistro and colleagues (2017) and Misra and colleagues (2014) used the micronucleus assay to assess potential mutagenicity of e-liquids. The micronucleus assay detects clastogenic and aneugenic DNA damage that results in the disruption or breakage of chromosomes, leading to portions of the chromosome being added, deleted, or rearranged following cell division. Canistro and colleagues (2017) found an increase in the percentage of micronucleated reticulocytes (immature red blood cells) following in vivo exposure to e-liquids. In contrast, Misra and colleagues (2014) found no significant increase in Chinese hamster ovary (CHO) cells exposed to e-liquids for 24 hours, including cells exposed to tobacco smoke extract. Again, although the same endpoint assay was used in each of these studies, the differences in outcomes may be due to substantial differences in experimental design, with the in vivo study of e-cigarette exposure finding a positive effect, whereas the study in a cell line exposed directly to e-liquids had no effect.
Oxidative Damage to DNA (8-OHdG Formation)
Canistro and colleagues (2017) evaluated two measures of oxidative stress following in vivo, whole-body exposure of male Sprague-Dawley rats to the equivalent of 1 ml/day of e-liquid containing 18 mg/ml of nicotine in an inhalation chamber. Levels of 8-hydroxy-2′-deoxyguanosine (8-OHdG) in the lung, and the reducing power of the lung tissue (ferric reducing antioxidant power, or FRAP) were assessed in the animals after 4 consecutive weeks of exposure via a smoking machine (11 17-second puff cycles per day, 5 days per week). The formation of 8-OHdG is a widely used biomarker of oxidative damage to DNA. It has been associated with increased mutagenesis in a number of test systems, and is often considered as an intermediate biomarker of carcinogenic potential (Curtin, 2012; Kasai, 1997). They found a statistically significant, approximately fourfold increase in the levels of 8-OHdG in the lung tissue of exposed rats. There was a strong inverse correlation between the FRAP activity in lung tissue and 8-OHdG levels (r = 0.845, n = 5), further supporting the conclusion that e-cigarette aerosols increase oxidative stress in the lung. The authors also measured the levels of antioxidant enzymes, including catalase, NQO1, superoxide dismutase, and glutathione S-transferase, and found a 25–35 percent decrease in activities in
all four enzymes. Because the expression of these enzymes is driven in large part by the levels of antioxidant “stress” through the Keap1-Nrf2/antioxidant response element, an increase in oxidative stress would normally be expected to increase, rather than decrease, the levels of expression of these enzymes’ pathway (Ma, 2013). It is not clear why the levels of these enzymes would be decreased, rather than increased, following exposures to e-cigarette aerosols that appear to be inducing oxidative stress in the lung.
Although oxidative damage to DNA is widely regarded as a potentially significant contributor to carcinogenesis, the vast majority of oxidative damage to DNA occurs via endogenous processes. The extent to which exogenous factors that induce oxidative stress and thus the formation of 8-OHdG contribute to actual tumor development is uncertain, and thus the utility of 8-OHdG to serve as a predictive biomarker of carcinogenesis is very limited.
Cell Transformation (Promoter Activity)
Although most in vitro assays focused on evaluating carcinogenic potential of xenobiotics use mutagenesis and/or oxidative free radical production, a few in vitro tests can assess the potential for a substance to act as a promoter of carcinogenesis. Breheny and colleagues (2017) used a cell transformation assay to assess the potential promoter activity of e-cigarette–generated ACM, and compared it to the promoter activity from tobacco smoke total particulate matter (TPM) collected from a reference combustible tobacco cigarette (3R4F). ACM was collected from a Vype ePen with an e-liquid cartridge containing blended tobacco flavor and 18 mg/ml of nicotine, using a linear smoking machine. Seven different ACM concentrations, ranging from 3 to 120 µg ACM/ml, were used in exposures to Bhas 42 mouse fibroblasts. They found that TPM was positive in all of three experiments, whereas ACM was negative in all three experiments (see Table 10-1).
Relevance of DNA-Damage/Mutagenicity Studies and the Presence of DNA-Reactive Chemicals in E-Cigarette Aerosols to Potential Human Cancer Risk from E-Cigarette Use
As discussed above, some of the in vitro studies have found evidence that chemical constituents of e-cigarette aerosols are capable of reacting with DNA and in some instances inducing mutations in vitro and following in vivo exposure (Canistro et al., 2017). Some of the chemical constituents found in e-cigarette aerosols, including especially the reactive aldehydes formaldehyde and acrolein, are DNA-reactive, and
formaldehyde has been shown to cause nasopharyngeal cancers in animals exposed via inhalation, and is considered by the Environmental Protection Agency and the International Agency for Research on Cancer to be a “known human carcinogen.” Although highly reactive with both protein and DNA, acrolein to date has not been shown to be carcinogenic in laboratory animals following long-term exposure via ingestion. However, no long-term inhalation studies in laboratory animals have been completed. As discussed in Chapter 5, formaldehyde and acrolein are present in e-cigarette aerosols. It should be noted that both of these reactive aldehydes are formed endogenously at low levels, and are present in many food items and may “off-gas” from commercial products, leading to frequent and widespread, but low-level, exposures to these compounds. It should also be noted that both acrolein and formaldehyde are highly reactive, and adverse health effects, including potentially cancer, are most likely to occur at the site of exposure (e.g., in the oral cavity and tracheobronchial tree). Cancer risk from environmental exposures to potentially DNA-reactive/mutagenic chemicals is a function of dose of the chemical at the target site. Using formaldehyde as an example, it is informative to put the levels of formaldehyde in e-cigarette vapors in perspective with other sources of exposure. As discussed in Chapter 18 (Harm Reduction), Goniewicz (2014) measured the levels of formaldehyde and other carbonyl compounds in e-cigarettes and compared them with the levels present in combustible tobacco cigarettes. When adjusted to “cigarette equivalents” (amount emitted in one combustible tobacco cigarette, and amount present in 15 “typical” puffs of an e-cigarette), the predicted exposures to both formaldehyde and acrolein were about 10- and 50-fold lower from e-cigarettes in most circumstances (see Table 10-2).
Gillman and colleagues (2016) did a similar comparison, using five different devices, each at four different “power” levels. Two of these devices generated remarkably high levels of both formaldehyde and acrolein—levels well above those found in cigarette smoke, and levels that exceed occupational standards for both of these substances (see Table 10-3). They noted that these two devices likely suffered from poor “wicking” of e-liquid to the heating element, which would generate much higher temperatures, facilitating decomposition of the humectant (PG) to carbonyls. They also noted that device 1 was not widely used, likely because of the frequency with which it would generate highly irritating aerosol (so-called “dry puff”). The other three devices generated levels of both formaldehyde and acrolein that were similar to those reported by Goniewicz and colleagues (2014).
Furthermore, it should be recognized that the contributions of formaldehyde, acrolein, and other reactive carbonyls present in cigarette smoke are likely relatively insignificant contributors to the known cancer risks
| Tobacco Smoke, 1 Cigarette (Roemer et al., 2004) | E-Cigarette Aerosols per 15 Puffs (Goniewicz et al., 2014) | |||
|---|---|---|---|---|
| Formaldehyde | Acrolein | Formaldehyde | Acrolein | |
| Mean | 25.39 | 53.88 | 2.83 | 1.15 |
| SD | 21.80 | 31.81 | 1.82 | 1.35 |
| Min | 3.70 | 15.50 | 0.32 | 0.01 |
| Max | 75.50 | 98.20 | 5.61 | 4.19 |
| Ratio cigarette/e-cigarette mean | 8.97 | 46.98 | ||
NOTE: SD = standard deviation.
SOURCES: Adapted from Roemer et al., 2004, and Goniewicz et al., 2014.
from combustible tobacco products. Thus, although there is substantial evidence that these DNA-reactive and potentially mutagenic compounds are formed and present in e-cigarette aerosols, the relatively low levels of exposure, coupled with the relatively low carcinogenic potential of these compounds, suggest that cancer risk from long-term use of e-cigarettes, if any, is likely to be very low, when compared with that from combustible tobacco cigarettes.
STUDIES OF EFFECTS OF MAJOR COMPONENTS OF E-CIGARETTES ON CANCER OUTCOMES
As described earlier regarding optimal study design, given the relatively recent introduction of e-cigarettes, there is a paucity of evidence on the long-term effects of e-cigarettes on cancer outcomes. Consequently, the committee drew on existing evidence of major components of e-cigarettes—namely, nicotine and the humectants PG and glycerol—with an emphasis on cancer outcomes. The committee discusses findings from epidemiological and in vivo animal studies in this section, from which the committee could draw inferences about the potential carcinogenic effects of e-cigarettes, but the discussion does not represent results of a systematic review.
| Power Level | Units Adjusted to µg/15 Puffs | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Device 1 | Device 2 | Device 3 | Device 4 | Device 5 | ||||||
| Form. | Acro. | Form. | Acro. | Form. | Acro. | Form. | Acro. | Form. | Acro. | |
| P1 | 128 | 104 | 4 | 0.9 | 1.1 | 0.6 | 2.0 | 0.8 | 2.0 | 1.2 |
| P2 | 315 | 255 | 23 | 5.0 | 1.1 | 0.9 | 4.2 | 0.9 | 3.2 | 2.4 |
| P3 | 480 | 375 | 120 | 39.0 | 0.8 | 0.5 | 2.1 | 0.8 | 4.7 | 2.3 |
| P4 | 765 | 615 | 255 | 124.5 | 8.9 | 8.0 | 3.2 | 0.9 | 5.1 | 2.4 |
| Mean | 422 | 337 | 100 | 42.3 | 2.9 | 2.5 | 2.9 | 0.8 | 3.7 | 2.1 |
| SD | 270 | 216 | 115 | 57.4 | 4.0 | 3.7 | 1.0 | 0.1 | 1.4 | 0.6 |
| Min | 128 | 104 | 4 | 0.9 | 0.8 | 0.5 | 2.0 | 0.8 | 2.0 | 1.2 |
| Max | 765 | 615 | 255 | 124.5 | 8.9 | 8.0 | 4.2 | 0.9 | 5.1 | 2.4 |
| Ratio | 0.1 | 0.2 | 0.3 | 1.3 | 8.7 | 21.8 | 8.9 | 65.3 | 6.8 | 26.1 |
NOTE: Acro. = acrolein; Form. = formaldehyde; SD = standard deviation.
SOURCE: Adapted from Gillman et al., 2016.
Epidemiological Studies
As reviewed below and elsewhere (Grando, 2014; HHS, 2014; IARC, 2000; Shields, 2011), the potential carcinogenicity of nicotine has been studied extensively in the in vitro and in vivo settings. However, there is a paucity of epidemiological evidence assessing the potential association between nicotine per se and the risk of cancer in humans. This is largely because studying the potential association between nicotine exposure and human cancer poses methodological challenges that severely compromise the generation of meaningful data. This is because prior to the advent of e-cigarettes, in recent decades the “purest” form of nicotine exposure has been via nicotine replacement therapies (NRTs). Given that these are smoking cessation medications, teasing an isolated contribution of NRTs in relation to cancer risk in the context of extensive prior/current combustible tobacco cigarette smoking histories is complex. For example, among smokers the overall contribution of nicotine exposure from NRT can only be expected to be a very small fraction of a smoker’s overall nicotine exposure because it will be greatly outweighed by the nicotine exposure from years of smoking combustible tobacco cigarettes. Continued greater use of NRT usually occurs in more addicted smokers who have a more difficult time quitting (Alberg et al., 2005), introducing the potential for strong confounding.
With these inherent challenges, the Lung Health Study provides the highest quality evidence on this topic to date (Murray et al., 2009). The advantages of the Lung Health Study are that it was a smoking cessation trial that tested an NRT (nicotine gum) and thus had detailed NRT use and combustible tobacco cigarette smoking data in the intervention group for a period of 5 years. In this study 3,320 participants from this intervention group were followed up for an additional 7.5 years to ascertain cancer outcomes, thus providing evidence on this topic from a prospective cohort study that emanated from the original randomized trial. Despite these strengths, in addition to the generic limitations noted above are limitations introduced by the fact that the cohort size and duration of follow-up are limited for yielding adequate statistical precision, the nicotine doses from nicotine gum are small, and the NRT exposure use and assessment occurred so near in time to the follow-up for cancer outcomes that the study could only be expected to detect contributions that occur in the latter stages of carcinogenesis. Thus, the inferences from the study results that indicated no statistically significant or clinically meaningful increased risk of lung cancer, gastrointestinal cancer, or all cancers are twofold: (1) it is unlikely there is a strong association between NRT use and cancer risk in the short term, and (2) the evidence provided by this null finding does not rule out the possibility of a weaker association between nicotine and cancer in the short term.
No epidemiological studies have addressed the long-term health consequences, including cancer, of propylene glycol and glycerol. Despite the fact that propylene glycol has been widely used in theatrical settings and in a few other occupations (see Chapter 5), the absence of evidence on cancer related to this topic is demonstrated by the fact that the Department of Health and Human Services, the International Agency for Research on Cancer, and the Environmental Protection Agency have yet to classify the carcinogenicity of propylene glycol in humans.
In Vivo Animal Studies
Typically, rodent bioassays for carcinogenesis involve 2 years of continuous exposures, and no studies of this nature have been identified in rats, mice, or other laboratory animals. However, N’-nitrosonornicotine (NNN) and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), derived from the tobacco leaves, formed during the tobacco curing process, and reported in e-liquids and aerosol, may contribute to the overall carcinogenic activity of tobacco products. As noted previously, however, because the nicotine in e-cigarette liquids is not extracted from cured tobacco leaves, where NNN and NNK are formed, the levels of these potent mutagens in e-cigarette aerosols are extremely low compared with tobacco smoke. Nicotine itself, as used in nicotine replacement therapies, is challenging to study in relation to cancer risk in epidemiological studies; the one high-quality study to evaluate the potential carcinogenicity of nicotine in NRT, the Lung Health Study, yielded null findings (see Murray et al., 2009, above).
Several studies have evaluated the consequences of long-term exposure to nicotine in animal models (Haussmann and Fariss, 2016). Two lifetime (2-year) bioassays evaluating the carcinogenicity of nicotine have been completed. Waldum and colleagues (1996) conducted a 2-year inhalation exposure study of nicotine using young adult female rats exposed to a constant concentration (501 ± 151 µg/m3) of nicotine. Although more animals had tumors in the nicotine-exposed group (21/59; 36 percent) than the controls (6/25; 24 percent) (see Table 10-4), the types of tumors found in the exposed group were common in this strain of rat. The authors concluded that there were no “tumorigenic effects of nicotine on any organ in the body” (Waldum et al., 1996, p. 1345), although they did note that tumors in the pituitary gland (adenoma) were seen only in the nicotine-treated animals, and noted that nicotine has been shown to have “neuroendocrine actions” (Waldum et al., 1996, p. 1345).
Toth (1982) evaluated the potential carcinogenic effects of lifetime exposure of 0.5 or 0.7 mg/ml of nicotine, administered in drinking water for 24 months to groups of male and female Swiss mice. These concentrations translate to an average nicotine dose of approximately 150
| Tumors (Site and Type) | Nicotine Exposed (%) | Controls (%) |
|---|---|---|
| Mammary gland | ||
| Fibroadenoma | 15 | 24 |
| Adenocarcinoma | 2 | 0 |
| Pituitary gland | ||
| Adenoma | 7 | 0 |
| Atypical adenoma | 2 | 0 |
| Ovary | ||
| Granulosa-theca cell tumor | 2 | 0 |
| Adenocarcinoma | 3 | 0 |
| Skin | ||
| Histiocytoma | 2 | 0 |
| Metastasis (origin unknown) | ||
| Liver | 2 | 0 |
| Abdominal cavity | 2 | 0 |
| Total percentage of rats with tumors | 36 (21/59) | 24 (6/25) |
NOTE: Percentages may not add up to 100 percent due to varied reporting, rounding, and missing data from source.
SOURCE: Adapted from Waldum et al., 1996.
mg/kg per day assuming a body weight of 25 g. Due to higher water consumption in the low-dose group, the daily dose per mouse at the 0.5 mg/ml concentration was only slightly smaller than in the 0.7 mg/ml group. Although this was generally a well-designed study with a reasonably large sample size, it could be argued that the highest dose was not sufficiently high, as there were no indications of toxicity, and no impact on body weight development or survival, so it would not be defined as a “maximal tolerated dose,” which is often an expectation in carcinogenicity bioassays. A thorough histopathological examination was completed. The authors reported no increase in tumor incidence in either nicotine-exposed group. The author of the study concluded that nicotine was “not carcinogenic under the experimental conditions” (Haussmann and Fariss, 2016, p. 712; Toth, 1982, p. 72).
Murphy and colleagues (2011) evaluated whether nicotine administration could enhance the development of NNK-initiated lung tumors in A/J mice. They found that nicotine alone, administered at a daily dose of 0.15 mg/mouse for 46 weeks did not increase lung tumor multiplicity. NNK-treated mice had much higher lung tumor multiplicity compared
with controls, but administration of nicotine for 46 weeks had no significant effect on the multiplicity of lung tumors in mice.
Another recent in vivo study in rats investigated whether oral nicotine exposure could cause early histopathological changes in urinary bladder epithelium that might be consistent with early-stage bladder carcinogenesis. Dodmane and colleagues (2014) administered nicotine hydrogen tartrate to rats and mice at doses of 52 and 514 ppm, respectively, for 4 weeks in drinking water. They then did a detailed histological evaluation of the urothelial lining. They found histopathological changes (hyperplasia) in 70 percent of the rats and 40 percent of the mice, compared with none in control animals. They also found that rats had a non-significant increase in the mean BrdU labeling index relative to controls, although there was no evidence of cytotoxicity via scanning electron microscopy. The authors concluded that “these findings suggest that oral nicotine administration induced urothelial hyperplasia (increased cell proliferation), possibly due to a mitogenic effect of nicotine and/or its metabolites” (Dodmane et al., 2014, p. 49). They further hypothesized that such nicotine-induced urothelial cell proliferation could possibly “act synergistically with DNA adduct-forming aromatic amines to increase the incidence of tumor formation in the urinary bladder in tobacco users” (Dodmane et al., 2014, p. 53).
VULNERABLE/SUSCEPTIBLE POPULATIONS
Population characteristics that identify subgroups that bear a disproportionate burden of cancer in the United States are race/ethnicity, sex, and socioeconomic status. Among these, the groups with the highest cancer burden are African Americans, males, and those of lower socioeconomic status. When looking across these characteristics, African American males are a particularly high-risk group. Among African Americans, the use of NRT products has historically been very low (Fu et al., 2005, 2008; Trinidad et al., 2011), and the emerging surveillance data on e-cigarette use also indicate low prevalence of use by African Americans. The results of the literature search revealed that the few relevant human studies were largely or entirely carried out in predominantly white populations outside the United States. Future research that includes diverse U.S. populations will be essential.
Children and adolescents are also vulnerable populations. For lung cancer, the younger the age of initiation of combustible tobacco cigarette smoking, the greater the risk of developing lung cancer in adulthood even after adjusting for lifetime combustible tobacco cigarette smoke exposure dose (HHS, 2014). This enhanced lung cancer risk associated with smoking at younger ages is hypothesized to be due to increased susceptibility
of the developing lung to carcinogens due to more rapidly dividing cells compared with mature lungs.
SYNTHESIS
A systematic review of the current body of evidence relevant to the potential association between electronic cigarette use and cancer leads to the clear conclusion that the present body of evidence is simply too sparse to permit meaningful inferences to be drawn about either cancer or intermediate cancer endpoints. Furthermore, the human studies published on cancer-related lines of inquiry to date are not only few in number, but have not had an optimal level of methodological rigor to permit drawing even preliminary inferences. The sparseness of the current evidence and the low quality of the human evidence on this topic preclude making any evidence-based conclusions about the potential association between e-cigarette use and risk of cancer in human populations.
Finding: There are no available epidemiological studies on the potential association between e-cigarette use and cancer in humans to make any conclusions. This holds true for comparisons of e-cigarette use compared with combustible tobacco cigarettes and e-cigarette use compared with no use of tobacco products.
Conclusion 10-1. There is no available evidence whether or not e-cigarette use is associated with intermediate cancer endpoints in humans. This holds true for e-cigarette use compared with use of combustible tobacco cigarettes and e-cigarette use compared with no use of tobacco products.
Conclusion 10-2. There is limited evidence from in vivo animal studies using intermediate biomarkers of cancer to support the hypothesis that long-term e-cigarette use could increase the risk of cancer; there is no available evidence from adequate long-term animal bioassays of e-cigarette aerosol exposures to inform cancer risk.
Conclusion 10-3. There is limited evidence that e-cigarette aerosol can be mutagenic or cause DNA damage in humans, animal models, and human cells in culture.
Conclusion 10-4. There is substantial evidence that some chemicals present in e-cigarette aerosols (e.g., formaldehyde, acrolein) are capable of causing DNA damage and mutagenesis. This supports the biological plausibility that long-term exposure to e-cigarette aerosols could
increase risk of cancer and adverse reproductive outcomes. Whether or not the levels of exposure are high enough to contribute to human carcinogenesis remains to be determined.
While evidence in humans for associations between e-cigarette use and cancer is extremely sparse, more abundant data have been generated in the in vitro and in vivo settings, including some positive studies and some negative studies on mutagenesis of e-cigarette components. Due to the mixed results across different experimental conditions and for different outcomes, clear, consistent signals have yet to be observed.
REFERENCES
Alberg, A. J., J. L. Patnaik, J. W. May, S. C. Hoffman, J. Gitchell, G. W. Comstock, and K. J. Helzlsouer. 2005. Nicotine replacement therapy use among a cohort of smokers. Journal of Addictive Diseases 24(1):101–113.
Bonassi, S., R. El-Zein, C. Bolognesi, and M. Fenech. 2011. Micronuclei frequency in peripheral blood lymphocytes and cancer risk: Evidence from human studies. Mutagenesis 26(1):93–100.
Breheny, D., O. Oke, K. Pant, and M. Gaça. 2017. Comparative tumor promotion assessment of e-cigarette and cigarettes using the in vitro Bhas 42 cell transformation assay. Environmental and Molecular Mutagenesis 58(4):190–198.
Canistro, D., F. Vivarelli, S. Cirillo, C. Babot Marquillas, A. Buschini, M. Lazzaretti, L. Marchi, V. Cardenia, M. T. Rodriguez-Estrada, M. Lodovici, C. Cipriani, A. Lorenzini, E. Croco, S. Marchionni, P. Franchi, M. Lucarini, V. Longo, C. M. Della Croce, A. Vornoli, A. Colacci, M. Vaccari, A. Sapone, and M. Paolini. 2017. E-cigarettes induce toxicological effects that can raise the cancer risk. Scientific Reports 7(1):2028.
Chen, J., C. Bullen, and K. Dirks. 2017. A comparative health risk assessment of electronic cigarettes and conventional cigarettes. International Journal of Environmental Research and Public Health 14(4):382.
Curtin, N. J. 2012. DNA repair dysregulation from cancer driver to therapeutic target. Nature Reviews Cancer 12(12):801–817.
Dodmane, P. R., L. L. Arnold, K. L. Pennington, and S. M. Cohen. 2014. Orally administered nicotine induces urothelial hyperplasia in rats and mice. Toxicology 315:49–54.
Farsalinos, K. E., and G. Romagna. 2013. Chronic idiopathic neutrophilia in a smoker, relieved after smoking cessation with the use of electronic cigarette: A case report. Clinical Medicine Insights: Case Reports 6:15–21.
Fenech, M., N. Holland, E. Zeiger, W. P. Chang, S. Burgaz, P. Thomas, C. Bolognesi, S. Knasmueller, M. Kirsch-Volders, and S. Bonassi. 2011. The HUMN and HUMNxL international collaboration projects on human micronucleus assays in lymphocytes and buccal cells—Past, present and future. Mutagenesis 26(1):239–245.
Flouris, A. D., K. P. Poulianiti, M. S. Chorti, A. Z. Jamurtas, D. Kouretas, E. O. Owolabi, M. N. Tzatzarakis, A. M. Tsatsakis, and Y. Koutedakis. 2012. Acute effects of electronic and tobacco cigarette smoking on complete blood count. Food and Chemical Toxicology 50(10):3600–3603.
Franco, T., S. Trapasso, L. Puzzo, and E. Allegra. 2016. Electronic cigarette: Role in the primary prevention of oral cavity cancer. Clinical Medicine Insights: Ear, Nose and Throat 9:7–12.
Fu, S. S., S. E. Sherman, E. M. Yano, M. van Ryn, A. B. Lanto, and A. M. Joseph. 2005. Ethnic disparities in the use of nicotine replacement therapy for smoking cessation in an equal access health care system. American Journal of Health Promotion 20(2):108–116.
Fu, S. S., M. M. Kodl, A. M. Joseph, D. K. Hatsukami, E. O. Johnson, N. Breslau, B. Wu, and L. Bierut. 2008. Racial/ethnic disparities in the use of nicotine replacement therapy and quit ratios in lifetime smokers aged 25–44. Cancer Epidemiology, Biomarkers & Prevention 17(7):1640–1647.
Gillman, I. G., K. A. Kistler, E. W. Stewart, and A. R. Paolantonio. 2016. Effect of variable power levels on the yield of total aerosol mass and formation of aldehydes in e-cigarette aerosols. Regulatory Toxicology and Pharmacology 75:58–65.
Goniewicz, M. L., J. Knysak, M. Gawron, L. Kosmider, A. Sobczak, J. Kurek, A. Prokopowicz, M. Jablonska-Czapla, C. Rosik-Dulewska, C. Havel, P. Jacob, and N. Benowitz. 2014. Levels of selected carcinogens and toxicants in vapour from electronic cigarettes. Tobacco Control 23(2):133–139.
Grando, S. A. 2014. Connections of nicotine to cancer. Nature Reviews Cancer 14(6):419–429.
Haussmann, H. J., and M. W. Fariss. 2016. Comprehensive review of epidemiological and animal studies on the potential carcinogenic effects of nicotine per se. Critical Reviews in Toxicology 46(8):701–734.
HHS (U.S. Department of Health and Human Services). 2004. The 2004 Surgeon General’s report on bone health and osteoporosis: What it means to you. Rockville, MD: Department of Health and Human Services, U.S. Public Health Service.
HHS. 2006. The health consequences of involuntary exposure to tobacco smoke: A report of the Surgeon General. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health.
HHS. 2010. A report of the Surgeon General: How tobacco smoke causes disease: What it means to you. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health.
HHS. 2014. The health consequences of smoking—50 years of progress: A report of the Surgeon General. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health.
IARC (International Agency for Research on Cancer). 2000. IARC monographs on the evaluation of carcinogenic risks to humans, Volume 77: Some industrial chemicals. http://publications.iarc.fr/Book-And-Report-Series/Iarc-Monographs-On-The-Evaluation-Of-Carcinogenic-Risks-To-Humans/Some-Industrial-Chemicals-2000 (accessed October 19, 2017).
IARC. 2012. Monographs: Tobacco smoking. http://monographs.iarc.fr/ENG/Monographs/vol100E/mono100E-6.pdf (accessed December 19, 2017).
Kasai, H. 1997. Analysis of a form of oxidative DNA damage, 8-hydroxy-2’-deoxyguanosine, as a marker of cellular oxidative stress during carcinogenesis. Mutation Research 387(3): 147–163.
Luzhna, L., P. Kathiria, and O. Kovalchuk. 2013. Micronuclei in genotoxicity assessment: From genetics to epigenetics and beyond. Frontiers in Genetics 4:131.
Ma, Q. 2013. Role of Nrf2 in oxidative stress and toxicity. Annual Review of Pharmacology and Toxicology 53:401–426.
Madsen, L. R., N. H. V. Krarup, T. K. Bergmann, S. Brentzen, S. Neghabat, L. Duval, and S. T. Knudsen. 2016. A cancer that went up in smoke: Pulmonary reaction to e-cigarettes imitating metastatic cancer. Chest 149(3):e65–e67.
Manzoli, L., M. E. Flacco, M. Ferrante, C. La Vecchia, R. Siliquini, W. Ricciardi, C. Marzuillo, P. Villari, and M. Fiore. 2017. Cohort study of electronic cigarette use: Effectiveness and safety at 24 months. Tobacco Control 26(3):284–292.
Misra, M., R. D. Leverette, B. T. Cooper, M. B. Bennett, and S. E. Brown. 2014. Comparative in vitro toxicity profile of electronic and tobacco cigarettes, smokeless tobacco and nicotine replacement therapy products: E-liquids, extracts and collected aerosols. International Journal of Environmental Research & Public Health 11(11):11325–11347.
Murphy, S. E., L. B. von Weymarn, M. M. Schutten, F. Kassie, and J. F. Modiano. 2011. Chronic nicotine consumption does not influence 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone induced lung tumorigenesis. Cancer Prevention Research 4(11):1752–1760.
Murray, R. P., J. E. Connett, and L. M. Zapawa. 2009. Does nicotine replacement therapy cause cancer? Evidence from the Lung Health Study. Nicotine & Tobacco Research 11(9):1076–1082.
NRC (National Research Council). 2014. Review of the formaldehyde assessment in the National Toxicology Program 12th report on carcinogens. Washington, DC: The National Academies Press.
Poulianiti, K., C. Karatzaferi, A. D. Flouris, I. G. Fatouros, Y. Koutedakis, and A. Z. Jamurtas. 2016. Antioxidant responses following active and passive smoking of tobacco and electronic cigarettes. Toxicology Mechanisms and Methods 26(6):455–461.
Roemer, E., R. Stabbert, K. Rustemeier, D. J. Veltel, T. J. Meisgen, W. Reininghaus, R. A. Carchman, C. L. Gaworski, and K. F. Podraza. 2004. Chemical composition, cytotoxicity and mutagenicity of smoke from US commercial and reference cigarettes smoked under two sets of machine smoking conditions. Toxicology 195(1):31–52.
Shields, P. G. 2011. Long-term nicotine replacement therapy: Cancer risk in context. Cancer Prevention Research 4(11):1719–1723.
Stephens, W. E. 2018. Comparing the cancer potencies of emissions from vapourised nicotine products including e-cigarettes with those of tobacco smoke. Tobacco Control 27(1):10.
Swenberg, J. A., K. Lu, B. C. Moeller, L. Gao, P. B. Upton, J. Nakamura, and T. B. Starr. 2011. Endogenous versus exogenous DNA adducts: Their role in carcinogenesis, epidemiology, and risk assessment. Toxicological Sciences 120(Supplement 1):S130–S145.
Thorne, D., I. Crooks, M. Hollings, A. Seymour, C. Meredith, and M. Gaça. 2016. The mutagenic assessment of an electronic-cigarette and reference cigarette smoke using the Ames assay in strains TA98 and TA100. Mutation Research/Genetic Toxicology and Environmental Mutagenesis 812:29–38.
Thorne, D., S. Larard, A. Baxter, C. Meredith, and M. Gaça. 2017. The comparative in vitro assessment of e-cigarette and cigarette smoke aerosols using the γH2AX assay and applied dose measurements. Toxicology Letters 265:170–178.
Toth, B. 1982. Effects of long term administration of nicotine hydrochloride and nicotinic acid in mice. Anticancer Research 2(1–2):71–73.
Trinidad, D. R., E. J. Pérez-Stable, M. M. White, S. L. Emery, and K. Messer. 2011. A nationwide analysis of US racial/ethnic disparities in smoking behaviors, smoking cessation, and cessation-related factors. American Journal of Public Health 101(4):699–706.
Waldum, H. L., O. G. Nilsen, T. Nilsen, H. Rorvik, V. Syversen, A. K. Sanvik, O. A. Haugen, S. H. Torp, and E. Brenna. 1996. Long-term effects of inhaled nicotine. Life Sciences 58(16):1339–1346.
Welz, C., M. Canis, S. Schwenk-Zieger, S. Becker, V. Stucke, F. Ihler, and P. Baumeister. 2016. Cytotoxic and genotoxic effects of electronic cigarette liquids on human mucosal tissue cultures of the oropharynx. Journal of Environmental Pathology, Toxicology and Oncology 35(4):343–354.
Yu, V., M. Rahimy, A. Korrapati, Y. Xuan, A. E. Zou, A. R. Krishnan, T. Tsui, J. A. Aguilera, S. Advani, L. E. Crotty Alexander, K. T. Brumund, J. Wang-Rodriguez, and W. M. Ongkeko. 2016. Electronic cigarettes induce DNA strand breaks and cell death independently of nicotine in cell lines. Oral Oncology 52:58–65.
























