
2
Addressing Nutrient Needs Due to Loss of Function in Genetic Diseases
Session 2 was moderated by Erin MacLeod, Director of Metabolic Nutrition at the Children’s National Health System and a Planning Committee member. In the first presentation, Denise Ney, Professor of Nutritional Sciences and Affiliate Faculty Waisman Center at the University of Wisconsin–Madison, described the basis of nutritional needs in the genetic disorder phenylketonuria (PKU). The next speaker was Marni Falk, Executive Director of the Mitochondrial Medicine Frontier Program at the Children’s Hospital of Philadelphia and Associate Professor in the Division of Human Genetics within the Department of Pediatrics at the University of Pennsylvania Perelman School of Medicine. Falk provided an overview of mitochondrial diseases and explained the nutritional challenges in managing them. The third presenter, Charles Venditti at the National Human Genome Research Institute, National Institutes of Health (NIH), discussed the contributions of nutrients in complex inborn errors of metabolism, using methylmalonic acidemia (MMA) as a case example. Rounding out the presentations was Sue Berry, Division Director for Genetics and Metabolism in the Department of Pediatrics at the University of Minnesota. Berry discussed some of the lessons learned from efforts at nutritional management of inborn errors of metabolism. The session concluded with a moderated panel discussion and questions and answers with workshop participants.
UNDERSTANDING THE BASIS OF NUTRITIONAL NEEDS IN PHENYLKETONURIA1
PKU is an autosomal recessive disease and its nutrient requirements are unique due to its genetic basis and inheritance, which affects requirements for phenylalanine (Phe) and tyrosine. It is caused by more than 800 mutations in the phenylalanine hydroxylase (PAH) gene. Ney opened her presentation by explaining that PKU is characterized by a deficiency in the conversion of Phe to tyrosine. It is managed with the low Phe diet. An individual with classical PKU often needs to restrict Phe intake to 300 to 500 milligrams per day, about 10 percent of typical intake. Tetrahydrobiopterin is an essential co-factor for PAH. In 2009, the U.S. Food and Drug Administration (FDA) approved a drug called Kuvan, which provides a synthetic form of the tetrahydrobiopterin co-factor. Up to 20 to 50 percent of individuals with PKU, who have some residual PAH activity, respond to this co-factor supplementation with a decrease in plasma Phe, which allows them to increase their dietary Phe intake.
Evidence suggests both causality and an intake–response relationship with respect to the toxicity of Phe in PKU. Phe intake that exceeds anabolic needs increases Phe concentrations in blood and brain, resulting in profound cognitive impairment if PKU is not treated with a low Phe diet started shortly after birth. Maternal PKU results in impaired fetal development and congenital anomalies if the mother does not strictly control her blood Phe levels during pregnancy. The biomarker that is used clinically is blood Phe concentration, a surrogate for brain Phe concentration.
A PKU scientific review conference held by NIH used a data analysis from the Agency for Healthcare Research and Quality to conclude that moderate evidence exists for a threshold effect that a blood Phe level of 400 micromolar or greater is associated with IQs less than 85 (Lindegren et al., 2012). The current recommendation in the United States is lifelong treatment of PKU, with a goal of maintaining blood Phe in the range of 120 to 360 micromolar.
Low Phe Diet
Ney described that the low Phe diet for PKU is adhered to fairly well in early childhood, when rapid development occurs. However, lifelong compliance is very challenging. The diet has the following components:
___________________
1 This section summarizes information presented by Denise Ney.
- Elimination of all high-protein foods. This means no animal foods or high-protein vegetable foods, such as seeds, nuts, and legumes.
- Protein substitution and micronutrient supplementation with medical foods. In 1958, the first low-Phe formula for PKU, Lofenalac, was introduced in the United States. Lofenalac is formulated from a casein hydrolysate. This was followed in 1972 by formulations comprising mixtures of amino acids leaving out Phe, and these were designated as medical foods in 1988. A second type of medical food, introduced in 2010, uses the peptide, glycomacropeptide (GMP), a 64-amino acid glycophosphopeptide. GMP is found in milk within the K-casein micelle and contains no Phe in its pure form.
The Genetic Metabolic Dietitians International Organization has developed dietary recommendations for PKU (Singh et al., 2016). The organization recommends a 20 to 50 percent higher protein intake when amino acid medical foods provide the primary source of protein, based on the body’s rapid absorption and oxidation of amino acids, resulting in reduced protein synthesis. Ney noted that improved growth in children with PKU who are fed protein at levels above the Recommended Dietary Allowance2 has been documented. This principle is relevant to gastrointestinal (GI) conditions where amino acid and peptide formulations are used.
Phe Tolerance or Minimum Phe Requirement
For an individual with classical PKU (i.e., a virtual absence of activity in the PAH gene), the Phe tolerance, or minimum Phe requirement, is the amount of Phe needed for protein synthesis to support growth and maintenance. Several studies have established the Phe requirements for infants from birth to 3 months. The first Collaborative Study, based on food records and multiple Phe determinations for blood, showed the requirements were 55 to 62 mg Phe/kg/d (Acosta et al., 1977). These findings paralleled those of Fomon and Filer, which showed 42 to 61 mg/Phe/kg/d are required to support the growth of healthy infants (Fomon and Filer, 1967). After 3 months of age, minimum Phe requirements are lower for PKU compared to the Dietary Reference Intakes (DRIs).3
In contrast, nutrient requirements for individuals older than 4 years with PKU are poorly determined. Several approaches are used to establish Phe tolerance for adults with classical PKU. Using intensive dietary
___________________
2 For DRI values, see http://nationalacademies.org/HMD/Activities/Nutrition/SummaryDRIs/DRI-Tables.aspx (accessed June 6, 2018).
3 For DRI values, see http://nationalacademies.org/HMD/Activities/Nutrition/SummaryDRIs/DRI-Tables.aspx (accessed June 6, 2018).
counseling, along with food records and blood Phe determinations over a period of several months, Phe tolerance can be increased from about 5 to about 8.5 milligrams per kilogram per day (MacLeod et al., 2009), about one-third the most current amino acid requirements of 25 milligrams of a combination of Phe and tyrosine (FAO/WHO/UNU Expert Consultation, 2007). Observations from clinical trials suggest that this low level of Phe intake can be difficult to maintain by adult individuals.
Tyrosine Requirements in Phenylketonuria
Tyrosine requirements in PKU are poorly understood. As the primary substrate for the synthesis of the catecholamine neurotransmitters, the low levels of tyrosine often seen in PKU patients is a concern. The response is to supplement their diet with tyrosine. Ney’s research has shown that the prebiotic GMP improves the bioavailability of tyrosine in PKU.
Ney went on to explain that despite almost 50 percent lower intake of tyrosine with GMP compared to amino acid medical foods, fasting plasma tyrosine levels are not different and within the normal range. These findings were reinforced by a 13-month study from Portugal that demonstrated increased mean blood tyrosine levels in study participants with PKU using daily servings of GMP medical foods (Pinto et al., 2017).
A metabolomics analysis revealed that the tyrosine is converted by the gut bacteria to potentially harmful compounds, in this case, tyramine and phenol sulfate, which are associated with headaches and renal toxicity, respectively (Ney et al., 2017). The use of pre- and probiotic supplements to decrease the gut synthesis of these renal toxins is a common element of disease management in people with chronic renal disease.
Phenylketonuria and Conditionally Essential Nutrients
PKU is likely associated with conditionally essential nutrients. People with PKU have low cholesterol levels, possibly as a result of lower cholesterol biosynthesis due to changes in metabolism associated with high blood levels of Phe. Inflammation and oxidative stress observed in PKU likely affect nutrient requirements. Lastly, low levels of several nutrients are observed in PKU, especially in children with the condition.
In addition, low bone mineral density and increased risk of fractures have emerged as complications of treated PKU, which may be associated with the high dietary acid load in amino acid medical foods. Studies in healthy individuals demonstrate that chronic ingestion of a high dietary acid load is bad for bone, which provides a very large bicarbonate reservoir and assists the kidneys in maintaining systemic pH homeostasis. This concept is especially important with age-related declines in kidney function.
Ney’s research group has confirmed these observations in a pilot cross-over trial of eight individuals with PKU where they determined that renal net acid excretion in 24-hour urine samples was threefold higher with ingestion of amino acid compared to GMP medical foods (Stroup et al., 2017). In addition, calcium excretion was significantly increased outside of the normal range compared to GMP with similar intake of calcium and bone-related nutrients.
Ney concluded her presentation with the following summary points:
- Nutrient requirements for protein, Phe, and tyrosine are unique in PKU.
- The principles of nutritional management of PKU have relevance to chronic disease, in particular, increased protein requirements with amino acid–based diets, as are often required in GI disorders.
- More needs to be learned about how the unique nutritional management approaches in conditions like PKU affect the gut microbiota, in particular, nutrient bioavailability, the synthesis of both beneficial metabolites, such as short-chain fatty acids, and also potentially harmful metabolites.
- The concept of conditionally essential nutrients in PKU, with altered metabolism and inflammation or oxidative stress, dietary acid load, and bone health, also has relevance for chronic conditions.
- Even though PKU is a mono-genetic disorder, it still has tremendous genetic diversity, which suggests substantial variation in nutrient requirements within the PKU population.
NUTRITIONAL INADEQUACIES IN MITOCHONDRIAL-ASSOCIATED METABOLIC DISORDERS4
Mitochondria are sub-cellular cytoplasmic organelles that arose about 2 billion years ago from purple sulfur cyanobacteria. They play a major role in nearly every metabolic pathway involved in oxidation of nutrients to permit cell growth and function. In addition to their major function as the energy “powerhouse” of the cell, Falk explained that mitochondria have central roles in many other essential activities, including calcium homeostasis and apoptosis.
Mitochondrial Disease
Falk stated that due to their broad range of functions, mitochondrial malfunction can cause symptoms in any organ at any age by any mode
___________________
4 This section summarizes information presented by Marni Falk.
of inheritance. This multiplicity of effects created a long-standing skepticism about mitochondrial disease, which is gradually disappearing. Many people have tried to identify a universal biomarker for mitochondrial disease, but unfortunately, no single biomarker exists because of the many different genes that can be involved in causing mitochondrial diseases.
Gene mutations that cause primary mitochondrial disease may occur both in the deoxyribonucleic acid (DNA) within the mitochondrion itself and in nuclear DNA. More than 20 novel gene disorders that are recognized to cause mitochondrial disease have been identified each year for the past decade. Although each individual gene cause is fairly uncommon, mitochondrial disease as a whole is the most common inborn error of metabolism, affecting at least 1 in 4,300 people across all ages.
Although it has been known since the 1950s that mitochondrial disease involves dysfunction in the energy-generating pathway that occurs within mitochondria, it is now recognized that mitochondrial disease may involve many additional processes and pathways occurring within these organelles (see Figure 2-1).
Clinical Features of Mitochondrial Disease
In describing mitochondrial disease, Falk noted that a wide range of organs may be affected, although most patients do have some form of neurologic and/or muscle involvement. Indeed, the disease can affect any one of the four nervous systems, that is, the central, peripheral, autonomic, and GI nervous systems. In addition to neurologic problems, whose onset may occur at any age and range from headaches and balance problems to Parkinson’s or developmental regression, eye problems such as drooping eyelid, eye muscle movement problems, and vision loss from retinal and/or optic nerve dysfunction are very common. Heart muscle or rhythm problems either as the sole feature or as part of a progressive multi-system array of problems that develop over time can also occur. Other common problems include fatigue, exercise intolerance, muscle weakness, developmental delay, sensorineural hearing loss, GI dysmotility, liver problems, kidney problems, bone marrow insufficiency, infertility, acute metabolic instability, and a host of endocrine problems.
Therapies for Mitochondrial Disease
No proven effective therapies or cures for mitochondrial disease are available, mostly because it consists of so many individually rare, highly heterogeneous disorders. Exercise, both aerobic and isotonic, has been shown for about a decade to have therapeutic value, but clarity on an optimal diet is lacking. So-called mitochondrial supplement cocktails
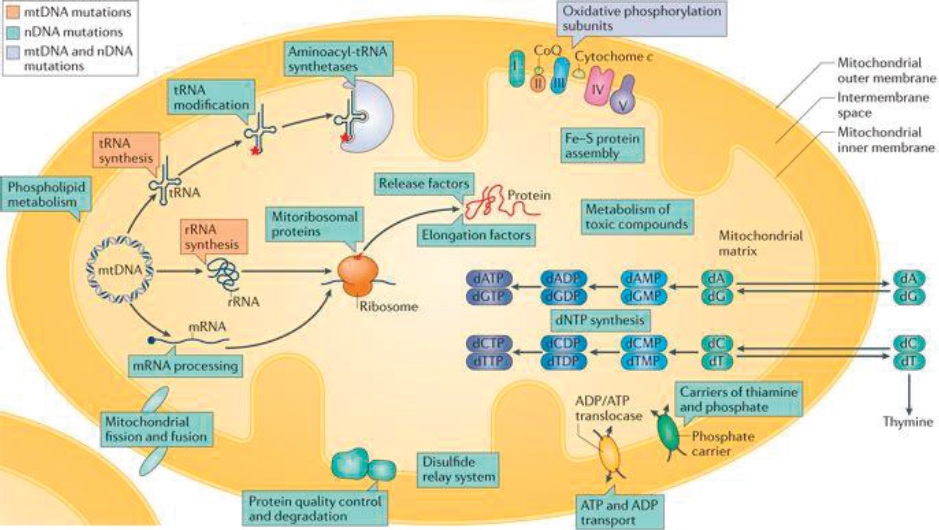
NOTE: ADP = adenosine diphosphate; ATP = adenosine triphosphate; DNA = deoxyribonucleic acid; Fe = iron; mRNA = messenger RNA; mtDNA = mitochondrial DNA; nDNA = nuclear DNA; rRNA = ribosomal RNA; S = sulfur; tRNA = transfer RNA.
SOURCES: As presented by Marni Falk, April 2, 2018; Gorman et al., 2016. Reprinted by permission from Springer Nature. Nature Reviews Disease Primers. Mitochondrial diseases, Gorman, G. S., P. F. Chinnery, S. DiMauro, M. Hirano, Y. Koga, R. McFarland, A. Suomalainen, D. R. Thorburn, M. Zeviani, and D. M. Turnbull, Copyright 2016. https://www.nature.com/nrdp (accessed June 14, 2018).
have been empirically used for some time based on the supposition they beneficially affect mitochondrial enzymes and cellular stress. These one-size-fits-all cocktails have great variability in their composition and clinical use, Falk stated. They commonly include
- supplements to increase free coenzyme Q pool (carnitine, pantothenate);
- enzyme co-factors (vitamins B1 or B2);
- metabolite therapies (arginine, folinic acid, creatine);
- enzyme activators (dicholoroacetate); and
- antioxidants (vitamins C or E, lipoic acid, coenzyme Q).
For the past decade, amino acid therapies have focused on arginine and citrulline. In some patients with mitochondrial disease, such as the common mitochondrial DNA syndrome called MELAS (mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes), acute stroke-like episodes have been shown to be mitigated with intravenous arginine. An expert consensus panel (Parikh et al., 2015) considered its use and determined that overall, it is well tolerated when administered at the proper doses and the patient is monitored for potential occurrence of low blood pressure and low blood sugar. A clinical trial is now under way to compare intravenous arginine and citrulline for treatment of acute stroke-like episodes in MELAS patients. Enteral arginine or citrulline is also commonly used as a prophylactic agent for acute metabolic stroke occurring in mitochondrial disease.
Clinical trials are now emerging, but as yet no universal clinical trial design, outcome measure, or biomarker has been established. Most of these trials have pursued antioxidant therapies for clinical syndromes, but without clear positive results. Newer agents are now under study in a range of clinical syndromes with a growing variety of therapeutic agents.
Nutritional Guidance in Mitochondrial Disease
To improve nutrition, many patients with mitochondrial disease may require a gastrostomy tube or parental nutrition, which is often associated with swallowing disorders, abnormal gut motility, and other GI complications. It is also known that essential micronutrient deficiencies may occur, including those of vitamin B12, vitamin D, folate, zinc, and others. Falk noted that multivitamin supplements safely alleviate some of these potential deficiencies, and that lutein is used in some cases if there is ophthalmologic involvement of the optic nerve or retina. However, she stated that very limited guidance is available to guide most aspects of nutrition in mitochondrial disease. No scientific data are available to
support guidance of optimal feeding intervals or specific macronutrient profiles involving the desired dietary ratio of proteins, carbohydrates, or fat. Although this issue has generated considerable discussion, current expert consensus recommendations do not include any clarity on the macronutrient profile. Current guidance suggests that energy, protein, and micronutrient intake should be evaluated and relative under-nutrition should be assessed in light of issues such as altered energy expenditures, abnormal intake, and absorption.
Because the glycolytic rate is typically increased in mitochondrial disease, high-carbohydrate diets may be beneficial. Furthermore, patients with acute metabolic stressors that may cause neurodevelopmental regression with decompensation, such as a fever or an infection, are often given glucose-containing fluids to prevent catabolism. Concerns have arisen about acute glucose infusion precipitating a metabolic crisis due to the increased nicotinamide adenine dinucleotide + hydrogen (NADH) to nicotinamide adenine dinucleotide (NAD) ratio that occurs in mitochondrial disease. A study being performed by Shana McCormack at the Children’s Hospital of Philadelphia’s Mitochondrial Medicine Frontier Program suggests that the type of carbohydrate may matter, as low-glycemic carbohydrates may offer a means to improve health outcomes and cognition in adult patients with genetically confirmed mitochondrial disease. Another concern is that diabetes mellitus is common in some patients. Falk concluded that even though glucose or low-glycemic carbohydrates might be possible therapies, much still needs to be learned about their optimization for acute and chronic care of patients with mitochondrial disease. The ketogenic diet (very high fat and very low carbohydrate) has been reported as a possible therapeutic approach for mitochondrial disease, particularly in patients with intractable epilepsy. By increasing ketones, succinate, and the starvation response, mitochondria biogenesis is increased and glutathione metabolism is enhanced. However, its clinical use is controversial due to inconclusive results from animal model studies. A major complicating factor is that the ketogenic diet is often not tolerated in patients because they metabolize fat poorly. Concerns also exist about the long-term health risks of the ketogenic diet. The modified Atkins diet, which involves a less extreme but still relatively high-fat and low-carbohydrate diet, has also been tried. A recent Finnish trial showed that although ten healthy adult control subjects had no problem completing a 4-week trial, all five mitochondrial myopathy adult subjects stopped the diet early due to severe muscle pain and burning as well as headaches and increased fatigue (Ahola et al., 2016). The explanation for this adverse response, stated Falk, was the impaired ability of mitochondrial myopathy study participants’ muscle fibers to burn fat aerobically
in mitochondria, as they have upregulated activity of their anaerobic glycolysis pathway to use sugar directly to generate energy.
Other nutritional recommendations include general common sense, such as having well-balanced diets with a range of fruits and vegetables, taking a multivitamin, avoiding fasting, and encouraging frequent small meals. In addition, good fluid intake is recommended, with increased fluid given when needed for heat, activity, or metabolic stress occurs.
Novel Treatment Strategies
Novel treatment strategies being considered to treat mitochondrial disease include genetic correction strategies as well as small molecular approaches, metabolic manipulation, and diet and exercise to treat the secondary cell consequences of the disease. Thinking about the mitochondrial respiratory chain as a “black box” factory that generates certain products, such as free radicals, adenosine triphosphate (ATP), nucleic acids, and nicotinamide dinucleotides, investigators are now looking at therapies that may not target the mitochondrial function proximally but, rather, focus on alleviating the resulting downstream deficiencies and broader cellular effects. The approach might involve activating certain signaling networks and biological processes, such as the mechanistic target of rapamycin complex 1 (mTORC1)-mediated control of cytosolic translation and autophagy. These processes are often dysregulated in mitochondrial disease, leading to a state of increased cellular stress. Falk showed a schematic illustrating this approach (see Figure 2-2).
Falk’s research laboratory group has tested more than three dozen antioxidants, metabolic modifiers, and signaling modifiers in a well-validated Caenorhabditis elegans (C. elegans) worm model of genetic-based primary mitochondrial disease. In each of these three treatment classes, some key therapies significantly restored the animals’ short lifespan. For example, lead signaling modifiers were nicotinic acid and probucol, and glucose was an effective metabolic therapy, as were the antioxidants N-acetylcysteine and vitamin E. The beneficial effects of each of these molecules when administered individually has been validated in vertebrate zebrafish animal models of primary mitochondrial disease.
Indeed, Falk’s group and many investigators around the world are evaluating different molecules and approaches (e.g., nutritional variation, drugs, and genetic therapies) in a wide variety of model animals and human cell types, testing their effects on biochemical as well as clinically relevant outcomes, such as survival, function, and feeling. Translational pre-clinical studies may also allow improved understanding of biomarkers for different molecular subtypes of mitochondrial disease. The results from this pre-clinical work have the potential to be used to devise a personalized
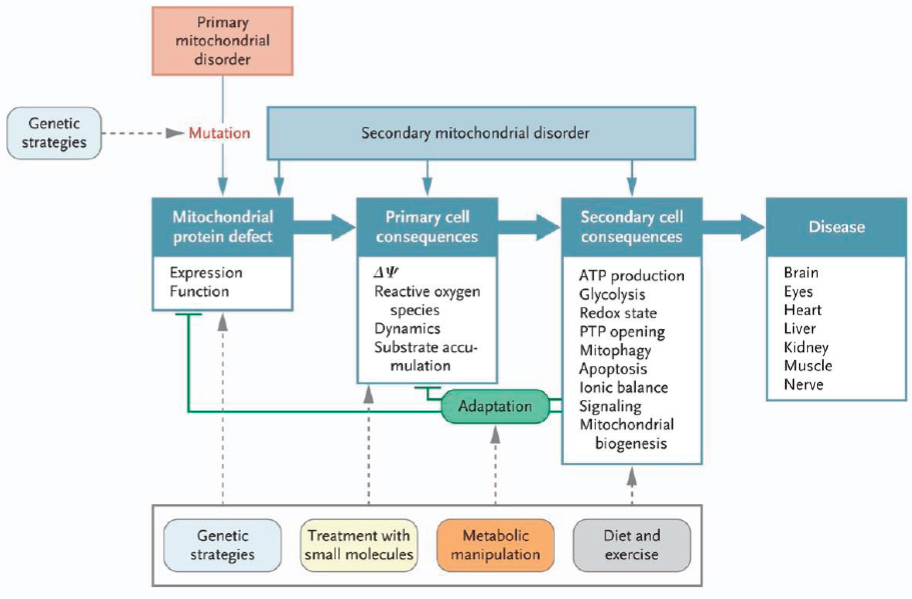
NOTE: ATP = adenosine triphosphate; PTP = permeability transition pore.
SOURCES: As presented by Marni Falk, April 2, 2018; Koopman et al., 2012. From the New England Journal of Medicine. Koopman, W. J., P. H. Willems, and J. A. Smeitink. Monogenic mitochondrial disorders. Volume 366, page 1139. Copyright 2012. Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
trial that objectively tests the therapy in a given patient or for a disease type. In Falk’s own research, some combined therapy combinations, or cocktails, appear to have synergistic effects.
Falk concluded with the following major summary points that reflect her own current thinking about therapeutic approaches to managing patients with mitochondrial disease:
- Some classical mitochondrial cocktail therapies do have objective therapeutic value in primary mitochondrial disease. However, their true value needs to be recognized as drugs intended to restore a state of health in genetically programmed mitochondrial diseases, rather than as unregulated dietary supplements that are intended for health optimization in the general, healthy population. Synergies between therapy components may be possible, if the proper doses, combinations, and unique mechanisms to treat different aspects of the disease pathophysiology can be identified.
- It is ideal to model therapies, when possible, in different mitochondrial disease subtypes before trying them empirically in clinic or prioritizing any one treatment randomly for further evaluation in a clinical treatment trial of mitochondrial disease patients. A promising diet to consider evaluating in primary mitochondrial disease patients may be one that is low glycemic and high carbohydrate rather than one that is high fat.
- It will be essential to evaluate the multiple potential clinical effects of dietary and nutrient therapies, to identify which aspects of these complex multi-systemic diseases respond and which clinical improvements are most valued by patients.
CONTRIBUTION OF NUTRIENTS IN COMPLEX INBORN ERRORS OF METABOLISM: THE CASE OF METHYLMALONIC ACIDEMIA5
MMA is a disorder of essential amino acid and odd chain fatty acid metabolism in humans. In essence, Venditti explained, four of the amino acids that are metabolized by the Krebs cycle—methionine, isoleucine, threonine, and valine, which are called propiogenic amino acids—cannot be oxidized because they lack their co-factor ado-cbl and instead they produce toxic acids in the body, such as methylmalonic acid. In MMA, the pathway to form ado-cbl from hydroxylcobalamin in the diet is aberrant. In the classical forms of MMA, patients have as much as 10,000 times the upper limit of normal methylmalonic acid in a healthy individual. MMA is the extreme end of a spectrum of metabolic perturbations related to
___________________
5 This section summarizes information presented by Charles Venditti.
vitamin B12 metabolism. MMA patients have a variety of symptoms and affected organs. All the patients eventually develop chronic renal failure and most patients have poor growth and obesity. The most worrisome aspect of MMA is that patients have a phenomenon of metabolic instability in which they can decompensate and die rapidly when they are subjected to stress, infection, or dietary indiscretion.
The definitive therapy for MMA, which is not yet available, is a gene or cell therapy that gives back the dysfunctional enzyme in every cell. The only real treatment that is offered to children with classical isolated MMA is an elective liver transplant procedure. A combined liver and kidney transplant is also done. Medical nutritional management with metabolites derived from valine, isoleucine, and odd chain fatty acids is used globally. Patients can also be given antibiotics to sterilize their GI tract to reduce the amount of propionate and in turn methylmalonic acid. An additional management option is to try to detoxify and relieve coenzyme A accretion by giving high doses of carnitine. Another approach, although its effectiveness is unknown, is to try to make the Krebs cycle work more efficiently by giving citrate. Mitochondrial target therapies can also be used in an effort to mitigate the electron transport chain effects.
Medical Nutrition Therapy to Treat Methylmalonic Acidemia
As mentioned, a frequent diet for MMA patients is a relatively low protein diet to restrict the propiogenic precursors, the amino acids valine, isoleucine, methionine, or threonine, and a medical food also without those amino acids (Manoli et al., 2016a,b). Venditti described a few studies that illustrated his concerns with management of these patients with medical foods that have not been designed appropriately. He explained an NIH study that followed a cohort of about 200 patients with various forms of MMA (www.ClinicalTrials.gov: NCT00078078) to understand patterns in the disease based on the subjects’ clinical phenotypes. Vendetti then presented data with results from this patient cohort, showing patient age and protein or equivalent, in grams per patient per day (see Figure 2-3).
The data show that some patients who were not hypermetabolic, and who should have been protein restricted, were in fact tolerating intakes far beyond the DRI6 (many of them at 200 percent of the DRI; see Figure 2-3). Venditti wondered whether the fact that these patients did not grow could be related to their diet, which had led to an imbalance in the metabolism of branched chain amino acids. As it turned out, the patients were prescribed medical foods formulas that contained four to
___________________
6 For DRI values, see http://nationalacademies.org/HMD/Activities/Nutrition/SummaryDRIs/DRI-Tables.aspx (accessed June 6, 2018).
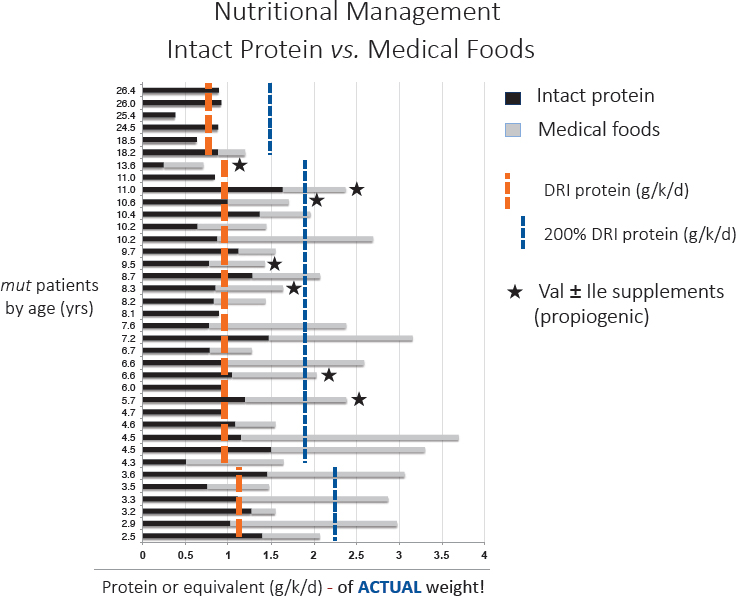
NOTES: The patients were given either an intact protein (black lines) or medical foods (gray lines). The DRI reference value and 200 percent of the reference value are provided as reference. Bars with a star * denote individuals who were given valine and isoleucine (propiogenic amino acids) as a supplement due to persistently low plasma amino acid concentration. DRI = Dietary Reference Intake; ile = isoleucine; val = valine.
SOURCES: As presented by Charles Venditti, April 2, 2018; Manoli et al., 2016a. Reprinted by permission from Springer Nature. Genetics in Medicine. A critical reappraisal of dietary practices in methylmalonic acidemia raises concerns about the safety of medical foods. Part 1: Isolated methylmalonic acidemias. Manoli, I., J. G. Myles, J. L. Sloan, O. A. Shchelochkov, and C. P. Venditti. Copyright 2015.
five times the DRI for leucine and zero isoleucine and valine (to prevent them from being propiogenic). However, Venditti’s analysis showed that the branched chain amino acid ratios in these patients were altered, probably because of the intake of medical foods. A further correlative analysis showed an inverse relationship between the level of medical foods intake and the levels of valine and isoleucine in these patients.
Furthermore, the more medical foods a patient took, the more aberrant the branch amino acids ratio became and the poorer the growth was.
A possible explanation is that the unbalanced leucine load in these patients likely promoted a decrease in isoleucine and valine. When the patient’s blood was too low in isoleucine and valine levels, an erroneous decision was made to supplement with isoleucine and valine. Valine and isoleucine are toxic precursors of methylmalonic acid. Venditti’s research group and other centers have been able to correct the branched chain amino acids deficiency syndromes in MMA patients by lowering the medical foods and lowering amino acid intake (Manoli et al., 2016a,b).
Venditti concluded his presentation with the following summary points:
- Medical foods are specially designed to treat patients with inborn errors of metabolism or other medical conditions, but too much can produce an iatrogenic toxicity syndrome in MMA.
- Reformulation of some of the medical foods and testing for effects on oxidation should be conducted to determine whether amounts given are perturbing amino acid levels.
- Research and other proof of concept studies are indicated for medical foods.
LESSONS LEARNED: WHAT IS KNOWN ABOUT NUTRITION MANAGEMENT FOR INBORN ERRORS OF METABOLISM7
As discussed in earlier presentations, inborn errors of metabolism are a large class of genetic disorders. They are typically single-gene abnormalities that are primarily nuclear and very few are maternally inherited. Essentially, the errors are failures to synthesize a product or a failure in catabolism, primarily due to single enzymes in those pathways, and are sometimes associated with enzyme co-factors, such as those in MMA. Elaborating, Berry explained that the primary focus of clinical attention is on the consequences of these errors in metabolism. These can be of several types, including
- decreases in product, such as in PKU;
- increases in precursors; this is true for PKU and even more so in MMA, where the tremendous increases in precursors can have toxic effects; and
- shunts to alternative pathways, as in orotic acid in urea cycle disorders; in some cases, these shunts can be used diagnostically.
___________________
7 This section summarizes information presented by Sue Berry.
Categories of Metabolic Diseases Where Nutrition Influences Care
Barry noted that metabolic diseases can also be grouped in terms of nutritional impact, including
- disorders of protein metabolism, such as PKU and MMA;
- disorders of carbohydrate metabolism, such as galactosemia;
- disorders of failure to generate glucose, such as glycogen storage diseases;
- fatty acid oxidation disorders, such as those involving failure to generate ketones; and
- mitochondrial disorders.
Treatment generally involves restricting or supplementing. Adequate calorie intake also needs to be ensured and this often involves providing substantial amounts of glucose. Monitoring nutrient adequacy is another key aspect of managing patients with these disorders. For example, clinicians must make sure protein is sufficient, but not excessive, in the protein disorders. Patients need to have enough essential fatty acids and may become very sick if fats are overly restricted in fatty acid oxidation disorders. Co-factor supplementation is another very important consideration in some cases. Scavenger therapies to get rid of undesired compounds have been used but need to be used carefully. Berry then suggested two additional strategies: monitoring toxic metabolites (i.e., keeping track of whether treatment is having the desired effect) and paying attention to biomarkers. Finally, clinicians must keep in mind that acute events inevitably occur, with various metabolic results.
Managing Inborn Errors of Metabolism
Reflecting on the preceding presentations in Session 2, Berry then described some of the issues that make the management of inborn errors of metabolism such a complex balancing act.
Phenylketonuria
In PKU, preventing the neurotoxic accumulation of Phe is the key management strategy. Some treatments, such as valine, isoleucine, and leucine, have been used to interfere with the transport of the amino acid Phe into the brain. In the early days of treating PKU, children died because all Phe was restricted; tyrosine then became a relative essential amino acid in that setting because Phe was restricted. This also proved that co-factors can be effective in some cases. Tetrahydrobiopterin, used
in treating PKU, is an example of how a co-factor can have a tremendous impact on care.
Berry added that the spectrum of the phenotype is very important in thinking about PKU as a paradigm disease. For example, the most severely affected patients with PKU will have a very limited tolerance of protein. In contrast, patients who have slightly milder mutations can be more generous in their protein consumption. This issue also highlights concerns about conditional essentially nutrients, especially in catabolic states. As PKU treatments have improved, clinicians have developed a better understanding of micronutrients that they previously did not consider. For example, initial issues encountered in selenium, zinc, and other deficiencies have now been resolved.
Mitochondrial Disorders
Both primary and secondary mitochondrial impacts are important in thinking about diet-related treatment approaches. No defined optimal diet exists for mitochondrial-associated disorders and few trials have examined this issue. The primary target for mitochondrial diseases is to maximize appropriate dietary components, including energy and micronutrients; avoid fasting and dehydration; and consider antioxidants. A limited number of mitochondrial disorders have a specific or diet co-factor need and in those cases, supplying these co-factors is very important.
Because the compounds used as primary intervention for mitochondrial diseases are currently being thought of more as treatments than as supplements, issues of access and availability need to be addressed.
Methylmalonic Acidemia
Berry stated that Venditti provided an excellent review of the importance of targeting medical foods to a disease so they do not result in amino acid imbalances. She noted that the lack of sufficiently large cohorts and the lack of financial resources to study these issues are difficult problems to resolve. In MMA, poor growth and obesity outcomes are related to the failure to understand the broad spectrum of nutritional needs in these children.
Comparing Inborn Errors of Metabolism and Normal Nutritional Needs
Berry reflected on clinicians’ responsibility to strike a balance in terms of nutrient needs in inherited metabolic diseases because the nutrient deficits are based on the individual disorder (e.g., tyrosine in PKU). The clinician’s job is to manage patients so they achieve intakes that are very
close to the required minimum; much still needs to be learned about how to strike this balance optimally. The use of artificial diets runs the risk of limiting micronutrients. Berry also reflected on the importance of the delivery method. For example, in the case of delivering amino acids, protein, or nitrogen equivalent, total parenteral nutrition is not an optimal way to give whole protein to patients.
Berry then described several key challenges in treating inborn errors of metabolism: (1) cost and access; (2) effects of treatments on the microbiome (the Urea Cycle Disease Consortium has a large project devoted to thinking about gut microbiome and its impact in the urea cycle disorder); (3) the need for medical foods to be optimally constituted and disorder-specific; and (4) the fact that nutritional needs change throughout the patient’s lifespan.
In terms of what is next in nutrition care for inborn errors of metabolism, she provided the following perspective:
- Huge gaps in information and knowledge still exist because of a dearth of longitudinal follow-up to determine natural history in inherited metabolic diseases, and a lack of clinical trials.
- Medical foods and supplements are not covered by insurance. This represents a tremendous burden on families.
- Research funding is desperately needed to determine whether some of these observations are generalizable to other rare disorders and micronutrient needs.
MODERATED PANEL DISCUSSION AND Q&A
In the discussion period following these presentations, participants addressed a variety of topics.
Efficacy of Alternative Medicine
The presenters were asked about the efficacy of traditional Chinese medicines for treating inherited metabolic disorders. Falk said that one of the limiting factors in studying these diseases is creating models and having well-defined and well-phenotyped patients. Advances in animal models are allowing researchers to systematically test potential compounds in high-throughput drug screens. In her opinion, over the next few years, systematic tests are needed of different compounds, different natural food products, and drugs to isolate the components that might have effects. Merely thinking about food or Eastern or Western medicine is not as helpful as thinking about the components, she said, and whether a component is really therapeutically effective.
Nutrients, Drugs, and the Regulatory Process
Several participants and panelists discussed the complexities of testing products that address distinctive nutritional requirements and how that intersects with the current regulatory structure for approving drugs for treatment. For example, no structure or patent protection exists to allow companies to develop drugs made from natural molecules. The current structure requires companies to file Investigational New Drug applications with FDA, which represents a risk for the companies.
Falk said it is a real worry that medical foods, which patients rely on to maintain their lives and improve their health, are not being regulated properly and resulting in unnecessary deaths on occasion. Collaboration is needed by all to create a better framework that results in well-regulated products that do not impose an undue financial burden on families. Falk said she thought it is important to change current thinking about how products are approved and how to define the populations for whom treatments are developed. For example, carnitine is an essential treatment in fatty acid oxidation diseases, but it is shown to lead to coronary artery disease in people in the general population who take it over time. The clinical and research communities need to better define the target populations for therapies, she said. Therapies need to be developed and regulated properly and made available to patients. Berry and Venditti both agreed with a participant’s comment about the value of medical foods and the need for research to improve them.
REFERENCES
Acosta, P. B., E. Wenz, and M. Williamson. 1977. Nutrient intake of treated infants with phenylketonuria. American Journal of Clinical Nutrition 30(2):198–208.
Ahola, S., M. Auranen, P. Isohanni, S. Niemisalo, N. Urho, J. Buzkova, V. Velagapudi, N. Lundbom, A. Hakkarainen, T. Muurinen, P. Piirila, K. H. Pietilainen, and A. Suomalainen. 2016. Modified Atkins diet induces subacute selective ragged-red-fiber lysis in mitochondrial myopathy patients. EMBO Molecular Medicine 8(11):1234–1247.
FAO/WHO/UNU (Food and Agriculture Organization of the United Nations/World Health Organization/United Nations University) Expert Consultation. 2007. Protein and amino acid requirements in human nutrition. Report of a joint FAO/WHO/UNU expert consultation. Geneva, Switzerland: World Health Organization. http://www.who.int/iris/handle/10665/43411 (accessed May 10, 2018).
Fomon, S. J., and L. J. Filer. 1967. Amino acid requirements for normal growth. In Amino acid metabolism and genetic variation, edited by W. L. Nyhan. New York: McGraw-Hill.
Gorman, G. S., P. F. Chinnery, S. DiMauro, M. Hirano, Y. Koga, R. McFarland, A. Suomalainen, D. R. Thorburn, M. Zeviani, and D. M. Turnbull. 2016. Mitochondrial diseases. Nature Reviews Disease Primers 2:16080.
Koopman, W. J., P. H. Willems, and J. A. Smeitink. 2012. Monogenic mitochondrial disorders. New England Journal of Medicine 366(12):1132–1141.
Lindegren, M. L., S. Krishnaswami, C. Fonnesbeck, T. Reimschisel, J. Fisher, K. Jackson, T. Shields, N. A. Sathe, and M. L. McPheeters. 2012. Adjuvant treatment for phenylketonuria (PKU). Comparative Effectiveness Reviews, No. 56. Rockville, MD: Agency for Healthcare Research and Quality.
MacLeod, E. L., S. T. Gleason, S. C. van Calcar, and D. M. Ney. 2009. Reassessment of phenylalanine tolerance in adults with phenylketonuria is needed as body mass changes. Molecular Genetics and Metabolism 98(4):331–337.
Manoli, I., J. G. Myles, J. L. Sloan, O. A. Shchelochkov, and C. P. Venditti. 2016a. A critical reappraisal of dietary practices in methylmalonic acidemia raises concerns about the safety of medical foods. Part 1: Isolated methylmalonic acidemias. Genetics in Medicine 18(4):386–395.
Manoli, I., J. G. Myles, J. L. Sloan, N. Carrillo-Carrasco, E. Morava, K. A. Strauss, H. Morton, and C. P. Venditti. 2016b. A critical reappraisal of dietary practices in methylmalonic acidemia raises concerns about the safety of medical foods. Part 2: Cobalamin C deficiency. Genetics in Medicine 18(4):396–404.
Ney, D. M., S. G. Murali, B. M. Stroup, N. Nair, E. A. Sawin, F. Rohr, and H. L. Levy. 2017. Metabolomic changes demonstrate reduced bioavailability of tyrosine and altered metabolism of tryptophan via the kynurenine pathway with ingestion of medical foods in phenylketonuria. Molecular Genetics and Metabolism 121(2):96–103.
Parikh, S., A. Goldstein, M. K. Koenig, F. Scaglia, G. M. Enns, R. Saneto, I. Anselm, B. H. Cohen, M. J. Falk, C. Greene, A. L. Gropman, R. Haas, M. Hirano, P. Morgan, K. Sims, M. Tarnopolsky, J. L. Van Hove, L. Wolfe, and S. DiMauro. 2015. Diagnosis and management of mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genetics in Medicine 17(9):689–701.
Pinto, A., M. F. Almeida, P. C. Ramos, S. Rocha, A. Guimas, R. Ribeiro, E. Martins, A. Bandeira, A. MacDonald, and J. C. Rocha. 2017. Nutritional status in patients with phenylketonuria using glycomacropeptide as their major protein source. European Journal of Clinical Nutrition 71(10):1230–1234.
Singh, R. H., A. C. Cunningham, S. Mofidi, T. D. Douglas, D. M. Frazier, D. G. Hook, L. Jeffers, H. McCune, K. D. Moseley, B. Ogata, S. Pendyal, J. Skrabal, P. L. Splett, A. Stembridge, A. Wessel, and F. Rohr. 2016. Updated, web-based nutrition management guideline for PKU: An evidence and consensus based approach. Molecular Genetics and Metabolism 118(2):72–83.
Stroup, B. M., E. A. Sawin, S. G. Murali, N. Binkley, K. E. Hansen, and D. M. Ney. 2017. Amino acid medical foods provide a high dietary acid load and increase urinary excretion of renal net acid, calcium, and magnesium compared with glycomacropeptide medical foods in phenylketonuria. Journal of Nutrition and Metabolism 2017:1909101. doi: 10.1155/2017/1909101.




















