
8
Large Databases and Consortia
Much of the power of today’s functional genomics depends on large databases that can hold the huge amounts of data that are being generated with a rapidly growing suite of technologies. However, there are a variety of challenges associated with developing and integrating such databases. One approach to creating and operating these large databases is through multi-institution consortia, which have their own challenges.
The workshop sessions described in this chapter examined some of the infrastructure challenges facing the field of functional genomics. In particular, the chapter combines two closely related sessions from the second day, one on the challenges and successes of integrating large datasets, and the other on the pros and cons of consortia and large databases.
The moderator of the first session, which was on large datasets, was Norbert Tavares of the Chan Zuckerberg Initiative. The speakers were Charles Danko of Cornell University, Alexis Battle of Johns Hopkins University, Rahul Satija of the New York Genome Center, Saurabh Sinha of the University of Illinois at Urbana-Champaign, and Genevieve Haliburton of the Chan Zuckerberg Initiative. The second session, which focused more on consortia, had no presenters but instead was made up entirely of a panel discussion.
CHRO-SEQ: A NEW TECHNIQUE FOR INTERPRETING GENOME SEQUENCE
The large datasets that are at the heart of an increasingly large portion of functional genomics are populated by a variety of techniques that produce an array of different types of genomic data. In his presentation,
Charles Danko described chromatin run-on and sequencing (ChRO-seq), a new technique that promises to provide an important window into the relationship between genotype and phenotype.
ChRO-seq is a way of measuring the genome-wide location and orientation of RNA polymerase, Danko explained, and he provided an illustration of what the resulting data look like (Chu et al., 2018; see Figure 8-1). The red bars pointing up denote the amount of RNA polymerase at each position in the genome going from left to right, while the blue bars pointing down denote transcription from right to left. These are the raw signal data, and a rich source of information about genome function, he said. “We can read off the location of enhancers and promoters, polyadenylation cleavage sites, and even these gene bodies just from the ChRO-seq data.” This is a good single assay for maximizing the information gained about the genome.
One way to think about the value of ChRO-seq is related to the fact that the amount of RNA polymerase loaded onto a gene corresponds extremely well with the levels of mRNA that are produced from that gene. “This goes along hand in hand with the idea that the majority [of the] variation in gene expression is regulated at the level of transcription,” Danko noted (Blumberg et al., 2019).
Additionally, the ChRO-seq data have features that correspond strongly with certain features of chromatin modifications. One of the major goals of his lab has been to “deconvolve what these chromatin states might look like.”
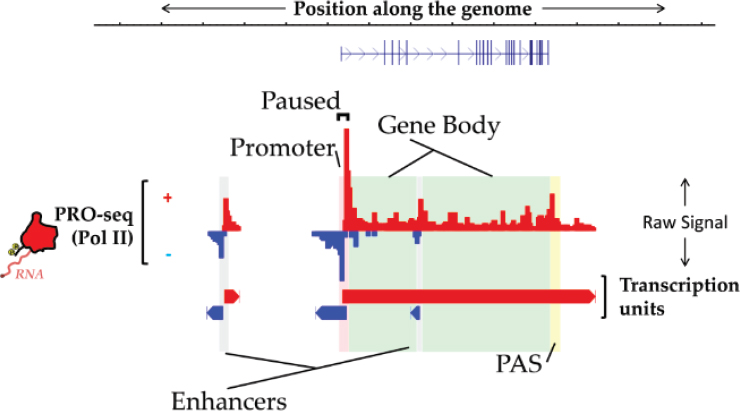
SOURCE: Charles Danko presentation, slide 2.
To accomplish this, the Danko lab took the ChRO-seq data and wrote a program to recognize the patterns of a number of different histone modifications. To this point, Danko said, they have trained models for 11 of the histone modifications, including H3K27 acetylation and H3K4 trimethylation, which are markers for promoters and enhancer regions; H3K36 trimethylation, which is a marker for gene bodies; and two markers for the repressed state. The only one of those that does not work at all is H3K9 trimethylation.
Using data from a stretch of chromosome 21 that was left out during the training of the model, Danko was able to test how well the model would impute distal marks along the chromosome by comparing the imputed values from the model against measured values from the Encyclopedia of DNA Elements (ENCODE) project. He displayed a couple of slides showing that the model closely reproduced both the broad patterns and the fine structure for most of the histone modifications.
One important question, Danko said, is how strong the relationship is between transcription and histone marks. “We’ve known for 60 years that transcription and histone modifications correlate with each other, but how strong is that correlation?” If there is a lot of unexplained variation, there could be multiple ways to encode a particular functional element in the chromatin state, and there are claims in the literature that this is the case. He asked, “Can we use this tool to evaluate whether those models are plausible or not?”
His team compared their own methods with chromatin immunoprecipitation sequencing (ChIP-seq) data that were downloaded from ENCODE. These comparisons were done on two widely used cell lines, K562 and GM12878. The imputations from their own data showed results similar to biological replicates of the downloaded ChIP-seq data.
However, Danko’s team found a number of cases where the imputations failed. In one, the imputation predicted a lot of H3K27 acetylation and there was no evidence for the same amount of acetylation in the ChIP-seq data from ENCODE. One possible explanation for this was the possibility of a great deal of biological variation between the K562 cells analyzed in ENCODE and those analyzed in his lab. Indeed, when they produced their own ChIP-seq data, that is what they found. There was clearly H3K27 acetylation there. This was similarly true for every one of the several hundred cases where their imputations differed substantially from the experimental ChIP-seq data. Because of this, Danko noted that “transcription is nearly identical to the histone modification for the marks they were studying.”
Another important question, said Danko, is whether the relationship between transcription and each histone modification varies with cell type. They compared models trained on K562 cells with data from a variety of
cell types including HeLa, T-cells, liver cells, mouse embryonic stem cells, and others. In every case except the mouse embryonic stem cells, “the active histone modifications were recovered with the same fidelity in the training cell type as in the other cell types, which indicates that the there’s extensive conservation in the relationship between these marks and transcription.” In the case of the one exception, Danko said he suspects that there is some fundamental shift in the association between the chromatin mark and transcription so that the relationship differs between embryonic stem cells and a fully differentiated cell.
Next, Danko asked, “Is transcription informative about complex chromatin states consisting of multiple different histones?” The classic example of such a complex state is a bivalent histone mark where H3K27 trimethylation and H3K4 trimethylation are found on the same nucleosome simultaneously (Young et al., 2017). Running their analysis in the region surrounding the bivalent prox1 gene, they found that they could recover the H3K4 trimethylation signal, although it did not work as well for H3K27 trimethylation. However, the model recognized that prox1 has “a promoter that should have H3K27 trimethylation on it.” So the answer to the question is yes: transcription contains substantial information about even such complex chromatin states.
Danko offered one more question that ChRO-seq can address: Does transcription initiate at all open regions? Danko showed examples explaining that it is not true that any open chromatin region can initiate transcription, but rather it is confined to specific regions.
To sum up, Danko said that the imputation works well and offers opportunities in genome annotation. He mentioned earlier speakers who talked about the need to sample a lot of different tissues in multiple experimental conditions in order to obtain functional elements in a species. “ChRO-seq is one strategy where you can use a single assay to extrapolate that kind of information on chromatin state and get gene expression patterns as well,” he said. “We’re also using this to better understand what the histone modifications actually do. Now that we have a model, we can interpret it by perturbing the modification or transcription and asking how the system changes?”
He closed with a series of challenges for this work: First, cell types that people think of as static actually vary a lot. This can create problems when researchers do not take the presence of biological variation into account. Second, differences in genome structure between humans and other organisms lead to technical challenges. In Drosophila, for example, the genes are more tightly packed. The implication is that machine learning models will have to be trained separately for these species. Finally, it is not yet clear whether transcription or histone modifications are informative in all species. “It’s plausible that we could have types of enhancers that are not
marked by anything,” he said. “I think we need to understand the basic mechanisms by which gene regulation works in order to extrapolate across species.”
USING GENE EXPRESSION TO UNDERSTAND THE GENETICS OF DISEASE
At Johns Hopkins University, Alexis Battle has been turning to gene expression data in her quest to understand the genetic variation that is associated with disease. “The focus of my lab,” she said, “has basically been to identify the effects of non-coding and regulatory variation on the cell and ultimately on disease.” These effects can be complex and depend on the specific content and specific tissues in which they take place. To deal with that complexity she develops computational models to work with large-scale gene expression data. “We do a lot of work in novel methods development and machine learning and statistical modeling to try to make sense of these data.”
Most disease-associated variants in the human genome are non-coding, Battle noted, which makes it difficult to use genome-wide association studies (GWASs) to understand the disease mechanism or to design interventions. Thus, she typically uses large-scale expression quantitative trait locus (eQTL) studies to examine the association between the genotype at a genetic locus and the RNA expression levels for a specific gene.
In theory, she said, eQTLs can help in the interpretation of genetic variation in complex disease. The idea is that if there is a non-coding variant and it is not known what gene it affects, eQTL data can point to a gene, we can design a drug, and everything is solved. Of course, it is more complicated than that. Many factors can alter the effects of a genetic sequence variant, such as environment, sex, and cell type, which can affect such things as transcription factor abundance and epigenetic changes.
To address these issues Battle has become involved in the Genotype-Tissue Expression (GTEx) project, which is a database focused on tissue-specific gene expression data (GTEx Consortium, 2017; Aguet et al., 2019). There have been 948 donors, with whole-genome sequencing done for each. They have also provided up to 54 different types of tissues—not everyone is able to donate every tissue, Battle noted—on which RNA sequencing has been performed. The resulting dataset has been used to look at GWAS variance and to interpret disease-associated variation in terms of what genes and what tissues those variants seem to primarily affect.
Unfortunately, Battle said, data from GTEx and other large studies such as eQTLGen only solve half of the problem because only about half of the GWAS hits co-localize, that is, share a causal signal with eQTLs in some tissue. However, there are still useful insights to be gained. For instance,
of the half of the GWAS hits that co-localize with an eQTL, about half of those co-localize with a gene that is not the nearest gene.
For the half of GWAS hits that are not informed by the eQTL studies, Battle asked, “What are we missing?” One thing that is not adequately considered, she said, is that gene expression is a dynamic process. “The effects of genotype on your cells change throughout your lifetime, during development, during disease progression, during aging,” she said. “Your genetic variants are acting differently at these different stages.” But almost all eQTL datasets, especially the large cohorts, are done at a single point in time, usually in healthy adults. Static healthy adult data may not reflect genetic effects in diverse, disease-relevant contexts.
To examine the issue of dynamic conditions directly, they began with induced pluripotent stem cells (IPSCs) from 19 genetically diverse individuals. These cells were grown in the lab, and then caused to differentiate into cardiomyocytes. They collected RNA-sequencing (RNA-seq) at 16 different points in time to produce a total of about 300 RNA sequencing samples along with genotype data for each cell line. Analysis of those data showed that gene expression is affected by genetic variation in a dynamic way. They observed, for instance, that many eQTLs that were present in the stem cell state disappeared as the cells differentiated to cardiomyocytes, and vice versa. But perhaps most intriguing, she said, are cases where a genetic variant has an effect on gene expression at intermediate stages of differentiation. Even though this was a study with a small sample size on a single-cell type, she said, “we’re starting to see explanation of some GWAS hits that are not explained by static tissue.”
Next, Battle turned to the issue of the effects of rare variants, which, she said, are completely missed by the sorts of studies she had been describing with GWAS hits and eQTLs. Each individual has about 50,000 variants in its genome that appear with a minor allele frequency (MAF) of 0.01 or less, as well as a few thousand variants not seen in any existing study and not found in any datasets (Li et al., 2014, 2017). The effects of most of these rare variants are unknown. What is known is that about 8 percent of Americans have a rare genetic disorder, and for about half of those, the variant has not been identified. And, Battle added, “I think 8 percent is a wild underestimate.” This is because we only sequence the people who have severe conditions and miss the impact of most rare variants, even of large effect, on many phenotypes.
To address this, Battle begins with the assumption that a variant having a functional impact on an individual’s health should be disrupting something at a cellular level. So, it should be possible to look for individuals whose expression for a particular gene is far outside the standard population distribution for that gene. She described a method she has developed to find such outliers that is called “splicing outliers.” While gene expression is
basically a one-dimensional measurement—either up or down—splicing can vary in a multiple-dimensional environmental space across all the different combinations of splice junctions.
Working from the GTEx dataset, she selected whole-genome sequencing and RNA-seq data across multiple tissues for 714 individuals of European ancestry. The unfortunate current limitation to people with European ancestry, she explained, was because the patterns of rare variation differ across different populations. Then she carried out an analysis of which sorts of variants are near genes for which someone is an extreme expresser or near genes for which someone is an extreme under-expresser. They found that certain types of variants, such as duplication and some interesting splicing variants, are associated with over-expression, whereas others, such as deletions and frame shifts, are associated with under-expression.
Finally, Battle briefly described a machine learning method her lab developed to predict which variants are likely to have a functional effect on genes in particular individuals. This method, which they call “Watershed,” is trained in a completely unsupervised manner, she said. “It really vastly improves performance over using whole-genome sequencing alone,” she said. By adding RNA from personal transcriptomic data, it is possible to take any state-of-the-art method and make it “better at identifying variants that actually look functional.”
Watershed can be used to inform disease association for rare variants in large studies. The bottom line, she concluded, is that “gene expression is very helpful for interpreting disease variants.”
AN ATLAS OF ATLASES
To begin his presentation, Rahul Satija noted that in recent years the quantity and types of data available to researchers in functional genomics have grown dramatically. Technologies for single-cell RNA-seq have been massively scaled, for example, going from tens or hundreds of cells in a single experiment to hundreds of thousands of cells in a single day. And a variety of profiling technologies have joined RNA-seq, such as single-cell assay for transposase-accessible chromatin using sequencing (ATAC-seq) or cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq), which can measure both RNA and protein simultaneously in single cells. There is also spatially-resolved transcript amplicon readout mapping (STARmap), which can make spatially resolved transcriptomic measurements in tissue. Each of these technologies offers a unique perspective on cellular identity (Wang et al., 2018). “If you want to know which transcription factors are particular drivers of cellular maintenance or cell state, you may want to look at the chromatin. If you want to identify surface markers for enrichment, you might want to use CITE-seq (Stoeckius et al., 2017).
And if you want to understand how a cell’s neighbors and environment influence behavior, you might need spatially resolved measures.”
While individually powerful, what might happen if these methods could all be used together to inform each other? “The goal of my lab,” Satija said, “is to think about a way that we might be able to form an integrated analysis within and across all of these different datasets, so that instead of looking at these aspects of cellular identity one at a time, we can combine them together and perhaps come up with a more holistic view for what a cell is doing.” The experimental and computational methods they have developed for this task were the topic of Satija’s talk.
A few years ago, four different groups profiled the human pancreas with single-cell RNA-seq with the goal of discovering cell types in the pancreas. There were four separate experiments done with four different single-cell RNA-seq technologies at four different labs. “We thought it would be interesting if we could integrate and pull these datasets together,” Satija said, but when they tried a straightforward type of meta-analysis, the result was a mess. The cells ended up grouping both by their underlying biological states and by the technology used to measure them.
To avoid this problem, Satija looked for a way to align the datasets so that cells in the same biological state grouped together across experiments. He developed a process he called “single-cell RNA-seq data alignment.”
The datasets from the four different experiments were very different, and it was difficult to integrate them or compare the cells in one set with those in another. To deal with that issue, Satija’s idea was to look for shared sources of variation across the datasets. For example, the correlation between two particular genes was conserved from one dataset to another. That was not by accident, he said. “These genes are both markers of alpha cells, so as a result they’re biologically co-regulated, so this correlation should show up regardless of what technology we look at.” His group started identifying such shared sources of variation, and then used those shared sources of variation as a scaffold for alignment.
The method works as follows, Satija said, and to keep it simple he used two datasets in his example (see Figure 8-2). The data in the datasets need to be from a single cell, but they do not have to be single-cell RNA-seq. The datasets should share some of the same underlying biological cell populations, but there can be some populations that are present in one dataset but not in the other. The first step is to use a method called canonical correlation analysis to project cells into a common cellular space. The next step is to look for “mutual nearest neighbors.” For each cell in the dataset, one looks for the most similar cell in the other dataset, and then the process is repeated. The lines or connections between mutual nearest neighbors in the two datasets are called anchors (Stuart et al., 2019).
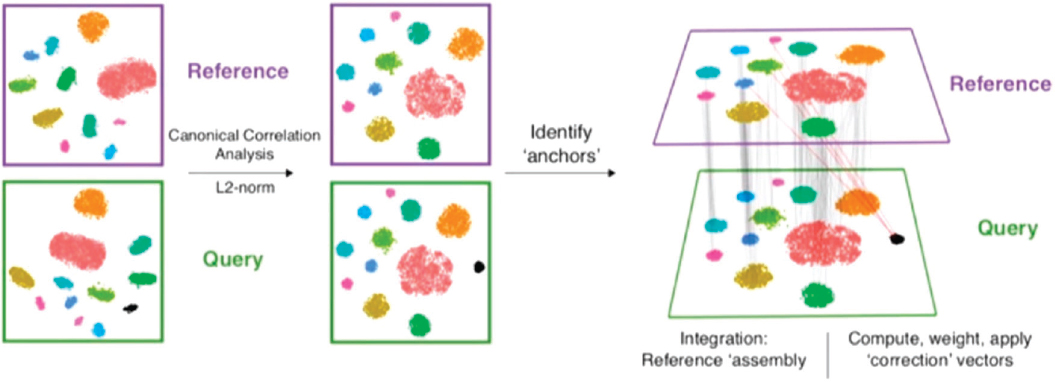
SOURCES: Rahul Satija presentation, slide 6; Stuart et al., 2019.
“The accuracy of these anchors is key,” Satija said, “because when we’re drawing an anchor, we’re making a biological statement that we think that two cells share some underlying biological similarity.” He referred audience members to his paper (Stuart et al., 2019) for the details on how to ensure that the anchors are correct, which he said is a “key part of the robustness of our method.” Once one has the anchors, it is relatively straightforward to pool the datasets into a harmonized reference set or to transfer information from one dataset to another.
Satija next described some of the ways this technique can be used. In one case his group went to the literature and found every single-cell RNA-seq set that had been published since the onset of the technology. Focusing on pancreatic islet cells, they integrated data from 25,000 cells collected with six different technologies from human and mouse subjects. The clusters that emerged were sharp and distinct. “By boosting our statistical power, we dramatically improve our ability to define and discover cellular phenotypes,” he said. One particularly impressive aspect of the work, he said, is the way that the datasets aligned across species. That opens a lot of possibilities.
In another example, researchers from the Allen Institute for Brain Science were interested in learning more about von Economo neurons, or spindle neurons, which are vulnerable in certain types of human diseases. Their function is poorly understood, and it is difficult to get healthy brain tissue from humans to study them. The researchers used Satija’s tool to align a single-nucleus RNA-seq dataset that they had generated from human brain tissue with a single-cell RNA-seq set they had previously generated in mouse. They found an exact match between the von Economo neurons and a particular cell type in mouse brains. Because there was something known about what that cell type does in mice, it offered some insights into the human von Economo neurons and opened the way to a series of experiments with which to learn more (Hodge et al., 2020). Satija noted, “his is an exciting opportunity in the same way that comparative genomics was essential for being able to interpret the genome.”
A more ambitious use of the alignment technique is to integrate datasets not just across an organ but across an entire organism. Satija’s group has done this with Tabula Muris datasets from the Chan Zuckerberg Biohub. The datasets were generated from 100,000 mouse cells from 26 different tissue types using two completely different technologies. Satija’s group was able to align the datasets, and through this process they were able to identify some “exquisitely rare populations” that made up only 0.001 percent of the entire dataset. These populations would have been undiscoverable if they had worked with the datasets independently.
There is no reason to be restricted to just single-cell RNA-seq data, Satija said. The same technique can be extended to single-cell ATAC-seq data and single-cell protein data, for example. He described a case using a
new technology that provides a spatial context as well. This new technology, STARmap, measures single-cell gene expression while keeping track of where each cell is located and, in fact, where each molecule is located, in three dimensions. The only downside to STARmap is that the limit is about 1,000 genes in a single experiment, he said, “which is pretty incredible, but it’s not transcriptome-wide like single-cell RNA-seq.”
His team set out to integrate STARmap (Wang et al., 2018) with single-cell RNA-seq (Tasic et al., 2018). By using the 1,000 genes from the STARmap spatial dataset to find anchors between it and the single-cell RNA-seq, they were able to effectively transfer information on all the rest of the genes that were not in the STARmap dataset and impute spatial gene expression from the single-cell RNA-seq work. After the analysis and validation, they were left with a transcriptome-wide spatial atlas of the mouse brain with “this beautiful gene expression measurement representing different layers in the cortex.”
Satija concluded by discussing two key uses for the integration technique that he has developed. One is to assemble a reference atlas. “If you have lots of different experiments produced across different labs, across different consortia, across different technologies … you want to build a single cellular atlas. These types of technologies can be used to build those references.” The assembly of such an atlas should be unsupervised, he said, and it will dramatically boost the statistical power to define rare and subtle cellular phenotypes.
The second is using the reference atlas to interpret new data. “Aligning datasets to that reference can help you to interpret information from other modalities, whether they’re coming from spatial measurements, or epigenomic measurements, or protein measurements,” he said. This alignment can also facilitate comparisons between different experimental models, such as aligning human data to mouse data, or to data from other species to perform comparative genomics.
A CLOUD-BASED PLATFORM FOR GENOMICS DATA MINING
The rate at which genomics data are generated is rapidly increasing each year, said Saurabh Sinha of the University of Illinois at Urbana-Champaign (UIUC), to the point that by the year 2025 there will be an astronomical amount of data to deal with. Because genomics is starting to generate much more data than astronomy, perhaps the adjective should be “genomical.”
Sinha mentioned that the National Institutes of Health’s (NIH’s) Genomic Data Commons project is one effort to deal with this issue of huge amounts of data. In particular, he highlighted two goals of that project:
- Developing and testing cloud-based platforms to store, manage, and interact with biomedical data and tools.
- Harnessing and further developing community-based tools and services that support interoperability between existing biomedical data and tool repositories as well as portability between service providers.
Those ambitions were the motivation for the establishment of KnowEnG, a center of excellence in big data computing, where he is a co-director and in charge of the data science research arm (Blatti et al., 2020). It is funded by NIH and involves researchers from UIUC and the Mayo Clinic. “Our goal was to build a cloud-based platform for genomics data mining,” he said, adding that software sharing and accessibility were two of the main foci.
Their idea at the highest level was that data analysis often involves a researcher having a spreadsheet of -omics data, which can be quite variable, and various forms of analysis can be applied to those data. The KnowEnG platform allows you to upload a spreadsheet and ask questions with machine learning and data mining tools.
In building the system, he and his colleagues thought about the important questions that people would want to address with the types of genomics data the portal was designed to support. The tools they included carry out tasks such as clustering of samples and gene set characterization, prioritization of the most important genes related to a phenotype, phenotype prediction from gene expression, and gene regulatory network reconstruction.
“One of the things we wanted these tools to do was to incorporate prior knowledge,” Sinha said. There are many public databases that provide extremely valuable information about genes, proteins, and their properties and relationships, and the people developing KnowEnG wanted the information in these databases to be available to inform the analysis of a user’s data. To do that, they captured all that information in a massive heterogeneous network, where the nodes were genes, proteins, and their properties, and the edges represented the relationships that these knowledge bases captured between nodes, such as protein–protein interactions, gene ontology information, pathways, and so on.
With the knowledge network established, the next task was to incorporate a user’s analysis. Suppose, for example, Sinha said, that a user has a spreadsheet of gene expression data and wants to do a classification task, perhaps identifying important genes. The knowledge network will use all its information about genes in carrying out the analysis of the information in the spreadsheet. “That’s our take on integrating large datasets,” Sinha said.
This approach posed two broad categories of challenges. The first set consisted of cyber infrastructure challenges. How can such a platform be made available for everyone to use? The solution they settled on was to put both the knowledge network and the tools to work with it on the cloud, where they would be in contact with each other. This had the advantages of being scalable and does not require any one lab to have the resources to support the knowledge base or the tools.
Building this cyber infrastructure involved “wrapping all the tools in containers so they are easily portable to a variety of different platforms and connecting those tools to storage buckets,” The end result, he said, is a cloud-based platform that has a Web portal where users can select a tool from the platform’s large collection, upload their spreadsheets, and have their data analyzed in the light of the large knowledge network.
One of the advantages of this approach, Sinha said, is that it is possible to link up with other resources on the cloud. For example, there is the Cancer Genomics Cloud, hosted by the same service, which has its own knowledge set and tools. KnowEnG and this platform are interoperable so that, for instance, a researcher working with data on the Cancer Genomics Cloud can also use tools on the KnowEnG knowledge network.
The other broad category of challenges facing KnowEnG, he said, relates to the algorithmic issue of how to incorporate the knowledge network into analysis. The major approach used by KnowEnG is based in network diffusion-based approaches. It is very popular in data mining, Sinha said, and is used by Google in its search engine operations.
As an example of how it works, Sinha discussed how the knowledge network had been used to guide the prioritization of genes determining drug response (Emad et al., 2017). The input was gene expression data from multiple cell lines, and the knowledge network was used to “smooth the gene expression data so that each gene’s expression not only reflects its measured expression, but also the activity or expression of its network neighbors.” The smoothed expression profiles of each of the cell lines were then correlated with drug response data on those cell lines to identify genes whose expression was most predictive of cytotoxicity in response to a particular drug. The smoothed expression profiles can be more powerful than the original expression profile because the smoothed profiles incorporate information from the prior knowledge network.
Sinha offered other examples of this knowledge-network-guided analysis, including a clustering of somatic mutation profiles of cancer patients. This can be difficult because collections of such mutation profiles are generally sparse. Using the knowledge network to smooth the profiles, he said, “can lead to much better detection of similarity among individuals and patients and much better clustering.”
In the future, Sinha said, there are still major unsolved problems, such as how to best determine which parts of the knowledge network to use in any given analysis, and how to deal with errors in the knowledge network. Another issue is that while network diffusion-based approaches have been shown to improve accuracy in many cases, they do not necessarily help reveal a mechanism. So, there is the question of how to use existing knowledge bases to make predictive models more mechanistic.
Sinha predicted that there will be more and more large and specialized databases in the cloud, and there will be a growing push for those databases to work together. There will also be a movement toward more automatic uses of knowledge bases by algorithms to analyze users’ data. The grand challenge, he said, is to take this to multi-level, mechanism-driven analysis of multi-omics datasets representing multiple scales of biological organization, informed by knowledge bases of molecular properties and interactions, and to be able to do it all on the cloud in a user-friendly way.
SUPPORTING DEVELOPMENT OF METHODS AND TOOLS
Genevieve Haliburton of the Chan Zuckerberg Initiative (CZI) described what the organization is doing to support biological research. Haliburton noted that she would not be presenting any original research because the initiative does not do research. Instead, she said, it attempts to find ways to support the development of methods and tools that can enable researchers such as those present at the workshop to do discovery research.
CZI funds three separate programs, one in science, one in education, and one in justice and opportunity. Because CZI is new, she had few outcomes to describe; instead, she spoke about what sorts of research and development are being funded.
The organization has two arms, a grant-making foundation and a technology organization, which have some overlap. The funding arm provides grants to individual researchers, groups of researchers, and existing standalone partners. One of its program areas is single-cell biology, which is where the organization engages with the functional genomics community. They fund a number of other project types, including open science, which is meant to foster healthy open research communities that really drive innovation. They support open access publication, open source software development, protocol sharing, and free and easy data sharing and reuse.
On the technology side the initiative has an in-house organization with designers, engineers, computational biologists, and others who build open-source software and tools aimed at enabling a wide range of sciences. Separate from the initiative is the Chan Zuckerberg Biohub, a medical science research center.
Haliburton listed several challenges that the initiative has heard about
from the functional genomics community and provided details on each. The challenges were
- protocol sharing and standardization,
- integrating data both within a modality and in a multimodal situation, and
- visualizing data for interpretation and collaboration.
The issue of protocol sharing and standardization arises both in the area of experimental methods and in computational tool development. Haliburton pointed to Protocols.io, which Steven Henikoff had mentioned in his talk the day before, as a platform for sharing protocols and as an example of methods sharing that the initiative would like to see more of. Open access to protocols allows for reproducible methods development, she said. In particular, the initiative has been funding the Human Cell Atlas (HCA) community, which now has hundreds of members, 125 publications, and more than 350 ongoing protocol discussions. These discussions are important for understanding how to perform a protocol. They also help to enable a global community of researchers.
The initiative has also funded grants for experimental and computational tool development related to the HCA work and, more recently, has funded collaborative networks in single-cell biology, most of them focused on single organs. The goal is to integrate and build a single network involving mathematicians, geneticists, tissue biologists, and clinicians all working together to provide a more comprehensive view similar to what is being done with the HCA.
The second challenge in functional genomics that Haliburton identified was data integration. “The way that I’m defining it right now is simply combining multiple studies for biological interpretation,” she said. This requires normalization, batch correction, and various other techniques, Haliburton said, and there are many people developing various algorithms in attempts to make this happen. Unfortunately, the algorithms are usually custom-made for a specific function. They are trying to find a more generalizable way to use the large amount of data that exists.
The technology side of CZI is helping develop the data coordination platform for the HCA, which is intended to be robust and scalable with a high-quality user interface, and that will house all of the data that are being generated. The goal is to build a platform that people can interact with on multiple levels, she said, “not necessarily just computational biologists but also people who have a hypothesis and want to go look at it.” Currently, the platform allows only downloads of individual study data, but there are plans to include standardized multi-study, multimodal integration.
Much of this learning is done through community working meetings,
Haliburton said. These meetings are called “jams.” Algorithm and method developers who have developed something with a specific purpose in mind but have encountered some challenges attend a jam, and others at the jam join in to help figure it out.
As an example, she described a “normjam,” where methods developers gathered to discuss high-level questions regarding normalization. “When I say normalizing here,” she explained, “I’m just talking about removing the technical variation from, say, a single-cell RNA-seq” in order to focus on the biological variation and not any variation due to differences in the equipment or methods.
Data integration methods inevitably involve trade-offs, Haliburton said. “You have to give up some information in order to align [different] various data types,” she said. “The more different your data are, the more you potentially have to give up. And if you know the downstream research question you want to be answering, you have some good idea of what you’re willing to give up and what you really need to hold on to.” However, she continued, if the goal is to come up with a method that is very generalizable for use in something like the HCA or that is interoperable between different datasets, it is not clear what trade-offs one might want to make. That is an area of active investigation that the CZI is supporting by engaging the community.
Haliburton’s last topic was visualization. “We know that a lot of our ability to interpret data and to infer things comes from looking at it from multiple perspectives,” she said, and one of the tools that the initiative has developed in-house is “cellxgene” (read as “cell by gene”). “It allows you to visualize any expression matrix or numerical matrix and explore it by a lot of different lines of metadata that you have,” she said. The metadata are a crucial tool, she added. For example, much of the work in the area is siloed, and in order to create bridges between these siloes, one needs good metadata describing the experiment in question. “All of the tools that we build are trying to deeply support a lot of different metadata.”
DISCUSSION
After these five presentations there was a short discussion on the challenges and successes of integrating large databases. Norbert Tavares, the manager of the single-cell biology program at CZI, started with a question. “As I’m managing this program, what do I need to look out for? What am I going to be blindsided by later?” More generally, what should funders be thinking about in this area?
Battle listed one of the challenges as the fact that the effects being studied are so highly context dependent. Collecting the data that will elucidate this context dependence will require huge sample sizes. It will be
necessary to think ahead about the effects one is looking for, how big they are likely to be, and what sorts of data are needed to answer the questions. Danko agreed and added that it is important to have people trained to be comfortable with both the biological and statistical sides of this work. Haliburton added that it is important to think about what one is considering as “phenotypes.” If one’s phenotype is just an increase in expression or a differential expression, it is difficult to make the connection between that and the real biological questions of interest. So, one challenge is to figure out how to make strong ties to the biology in these large abstract datasets.
Going in a different direction, Satija said that there is often a desire to automate analyses and have them all be done through a standardized pipeline. While it would indeed “be wonderful to just push a button and have all this magically happen, I feel like a lot of the steps that go into many of these approaches do require some biological supervision.” So when one is writing software, the goal should not be to write one piece of code that will be guaranteed to work for everybody, but rather to provide the appropriate amount of flexibility so that users can explore their data and return results that actually make biological sense.
An audience member commented that when researchers make their data available, it would be useful to have it as raw data. Battle commented that this is something that funders have a great deal of control over, because they can require researchers to deposit their raw data. Philip Benfey of Duke University asked about handling the sort of time-dependent data that are necessary to study the dynamics of the cell. “How do you analyze something that’s essentially four-dimensional?” Satija said he thought that one of the most exciting aspects of single-cell data is that if one is studying a developmental or transitioning process, even if the data seem to represent a static snapshot, there is still variation in those data in terms of exactly how far cells have moved along through the process. By combining a number of seemingly static snapshots, one can create datasets that include a fourth dimension, although that dimension is not time, per se, but rather “pseudo time” defined by the different points in the developmental progression. “We can order different events with extremely high precision and actually get to the point where we can almost understand causality or at least know that A comes before B, and therefore, B did not cause A.”
One of the first papers from his lab included data from an adult mouse brain and from a developing mouse brain, Satija said, “and the data from the developing mouse brain looked kind of like a cloud. It looked like the cells hadn’t differentiated yet.” So using the anchoring technique he described in his talk, his team found anchors between the developing cells and the differentiated cells, which allowed them to use the adult dataset, where there was plenty of diversity, to guide their analysis of the early embryo.
With that ability, they were able to see early hints of differentiation and ask, which were the first genes to change?
Gene Robinson of the University of Illinois at Urbana-Champaign asked those panel members working with big datasets about their thoughts on genomic security and privacy. “Are there safeguards that need to be put in place?”
Haliburton said that it is something she and her team have thought a lot about. “There are certain policy-driven limitations that are informing the way we’re thinking about it,” she said. In particular, they are building their platforms to support whatever sort of controlled access might be required by the consent forms that human subjects have signed. “Policy and science aren’t always aligned,” she said, “and the policy might not actually reflect what the science needs, so we’re trying to think deeply and thoughtfully about that because these things could get out of hand, and that would be very bad.”
Satija said that given how much personal information most people share on Facebook and other social media—information that can be much more sensitive than genetic information from a research study—it makes sense that when talking to the broader community about genetic privacy, we should not talk about it with fear. Putting genetic privacy in the context of other information is the goal we should be working toward.
Battle added that it is extremely important when getting consent from patients that they understand that “there are breaches of all of our sensitive data.” Researchers should do their best to protect people’s privacy, but they should also be clear that they cannot guarantee 100 percent certainty.
IMPORTANCE OF CONSORTIA AND LARGE DATABASES
The second part of the discussion focused on consortia and was moderated by Charles Danko of Cornell University. The discussion panel consisted of Felicity Jones of the Friedrich Miescher Laboratory of the Max Planck Society, Alexis Battle of Johns Hopkins University, Saurabh Sinja of UIUC, Rahul Satija of the New York Genome Center, and Sean Hanlon of the National Cancer Institute.
On the first day of the workshop, Aviv Regev offered her thoughts on the importance of large initiatives (see Chapter 2 for further description of Regev’s keynote address). Included in this was a brief overview of the HCA and other functional genomics initiatives.
People have always understood the value of maps, Regev said, so as more and more information was collected about human cells, the realization arose that it would be useful to have an atlas of human cells, and that realization led her and others to start the HCA initiative. The mission of
that initiative, she said, was “to create a comprehensive reference map of the types and properties of all human cells, the fundamental unit of life, as a basis for understanding, diagnosing, monitoring, and treating health and disease.” The originators of the initiative did not want to promise—or give people the impression that they were promising—that they would be curing all disease. “It’s a basic biology problem with basic biology answers. But it’s also useful, and that distinction needs to be made and repeated, especially in any public context.”
Because humans are diverse and so are their cells, Regev and her colleagues realized that no one lab or institute or country should build the atlas. Instead it should be an international effort that is open to everyone regardless of the funding source. “There’s over 1,700 members now in HCA from 71 countries and more than 1,000 institutes.” Openness is a core value of the atlas. The data coordination platform, the data releases, the lab protocols—everything the atlas does—is shared and in the open.
The first draft of the atlas is expected to span at least 100 million cells, including most major tissues and systems from healthy donors of both sexes with geographic and ethnic diversity and some age diversity. Ultimately, the comprehensive atlas is expected to have up to 10 billion cells representing all tissues, organs, and systems as well as full organs, again from a diverse group of healthy donors but also with mini-cohorts representing various disease conditions.
A sister initiative to the HCA is the International Common Disease Alliance (ICDA), which is focused on the connection between human genetic function and disease. A recent meeting developed a series of recommendations for the ICDA, which Regev reviewed. The first was to build reference atlases of tissues, diseases, and organisms.
A second line of recommendations involved performing massive high-content pooled screens for function. This will require continuing to develop new experimental modalities, as well as increasing efficiency and improving computational approaches.
A third set of recommendations centered on developing new module-level analytics for gene function that associate cell types with genes, use gene modules to understand biology and to increase signal detection, and detect interactions between single nucleotide polymorphisms both within and between gene modules.
Panel Discussion on the Pros and Cons of Consortia and Large Databases
On the second day of the workshop in the discussion period devoted to consortia such as the HCA, the participants talked about the advantages and the disadvantages of such consortia. Danko, the moderator, opened the
period by asking each of the panelists to describe whatever experience they had had working with consortia and what had worked and what had not.
Battle started by saying that the consortia she has primarily been involved with were the GTEx Project and also the HCA. She has mixed feelings about consortia, she said, but her work depends on very large datasets and those usually cannot be produced by a single lab. On the other hand, she said, about half of her lab’s projects are not working with large data, and she also does small one-on-one collaborations. She noted that these efforts “are often more well thought out in terms of looking for a very specific effect that we think is important.” So, both approaches—large consortia and smaller-scale research—can be valuable.
She added that her experiences with consortia have had some really good parts, and consortia helped launch her career. But there are also cases where people, especially students, get lost in the middle. One should also think about the effects that consortia have on training. Bottom line: there are pluses and minuses.
Sinha said that he had also seen positives and negatives from consortia. On the one hand they bring people together to share knowledge and learn from each other in unique ways. On the other hand, because consortium teams are larger, the chances of trainees getting lost are higher. Continuity can also be an issue. For example, if the consortium period ends, then it may be necessary to let go of the large software team that was assembled for the effort. For better or worse, the large datasets assembled by consortia can shape the direction of the science in strong ways.
Satija said that his best experiences with consortia have been in cases where there were small teams of people with complementary expertise working closely together. Some consortium grants explicitly require multiple principal investigators with complementary expertise. In his case there was a technology developer, an immunologist, and a computational biologist. On the flip side, it can be a problem in large consortia with groups that have complementary skill sets who are explicitly assigned different tasks. The groups may have been funded with separate applications and might be in very different parts of the country or world. If those groups are not tightly aligned, it can feel less efficient.
Jones said that her work with sticklebacks depends on having access to the datasets produced by large consortia, and that the stickleback community has received tremendous benefit from consortia, not just in the resources they make available but in terms of training as well. “We used to run a stickleback summer course where people from around the world would come and learn to do transgenics and bioinformatics, QTL [quantitative trait locus] mapping, and so on…. We have developed a really tight and friendly network of researchers in this area, and I think the stickleback community has really, really benefited from that.”
Hanlon said that the benefits of consortia include not only the data and tools that they generate but also the policies that they have instituted, such as pushing for early data sharing. In his experience, he said, consortia have been positive for trainees. “Many of the programs I’m involved in have some sort of junior investigator–associated meetings so we can have the trainees involved. Also, there’s a leadership opportunity many times for trainees. We have trainees that lead a number of the working groups and really contribute.”
An audience member suggested that with the costs of collecting genomics data dropping and the ease of collecting those data increasing, large consortia are likely to become less important. Battle agreed, but said that there are still some areas, particularly in human genetics, where the necessary datasets are too large for a single lab to collect at this time, “and if we are waiting for it to get cheap enough for one lab to do that or even have the time to do it, we would be waiting quite a long time.” Furthermore, a consortium like GTEx makes a major contribution by enforcing the uniformity of its data, which contributes to increased signal during processing.
Satija agreed with Battle and said that human genetics in particular is an area in which it is important to minimize technical variation in order to do extremely large-scale comparisons. Battle commented that the uniformity of data collection, often an advantage, can also be a disadvantage for consortia. “If a consortium like that makes the wrong choice, then you have spent a giant chunk of change on a technology that’s actually not the right one to use.” Danko noted that consortia can also have an impact by standardizing the methods of sample processing and what constitutes good data. An audience member asked how consortia could standardize data collection and analysis in the face of multiple conditions, for example, when different members of a consortium are working under different environmental conditions.
“I think that’s really challenging,” Battle said. Particularly as researchers begin to focus more on the environmental effects on human health, standardizing those measurements will be essential. She mentioned a study in which hospitals were given long and detailed instructions on how to collect each sample—exactly where to make a cut, for instance. “They were also extremely careful about recording everything…. It is beyond anything I’ve seen with other projects, and even then we still have huge sources of technical and experimental variation.” Central coordination can help, as well as standardizing protocols and sending training teams to help. “But regardless of how perfect you are, you’re still going to have huge sources of variation between sites.”
Hanlon commented that while it is often important to have standards across a consortium, it is a good idea to have some room for flexibility, and he offered an example of a study of tumors where it would have been
impossible to gather all of the required samples if doctors had to adhere to the original instructions.
Gary Churchill said that he appreciates consortia, not only because they are powerful mechanisms for generating large standardized datasets, which are extremely valuable, but also for how they—or at least GTEx—have made it easy for him to get information. “I go to the website. I type in the name of the gene that I’m interested in. I can immediately see which tissues it is expressed in and whether it has a QTL.”
Battle agreed and said that, more generally, it is valuable for consortia to have a user-friendly portal. When agencies fund large-scale data collection, she said, one requirement should be that the grantees make the data readily usable and easily accessible.
Hanlon added that the Human Tumor Atlas program was funded through the Cancer Moonshot Program, which has a goal of making data accessible to a broader community—not just biometricians, but also cancer biologists, and even patients and clinicians.
Marc Halfon of the University at Buffalo argued that the agencies who fund the development of databases should be ready to provide the necessary funding to maintain the databases over time. Technology can change, and data can become stale, so maintaining a database means more than just keeping it running as it has been for years.
Hanlon said that some federal agencies, including NIH, have programs aimed at maintaining databases. They are currently thinking about the broader issue of how to sustain them over time. Maintaining a project may actually be more difficult for smaller-scale efforts, Battle said. “Sharing data and maintaining data and maintaining methods and software [are] really challenging when it’s just your own lab.” For instance, when lab members move on, it can be difficult to interest incoming students or postdocs in maintaining a previous lab member’s software. “At least with consortia there’s usually a funding institution that cares that it’s maintained, and they will sometimes help,” she said.






















