
8
Electrochemical Engineering Approaches
8.1 OVERVIEW
Electrochemistry considers chemical reactions that result in the production or consumption of electricity. Technologies use electrochemistry to take measurements of chemical systems, use chemical reactions to generate electricity, or use electricity to drive chemical reactions. Several approaches have been proposed that use electricity to promote or to drive reactions that ultimately result in carbon dioxide (CO2) removal from the atmosphere (House et al., 2007; Rau, 2008; de Lannoy et al., 2012; Datta et al., 2013; Rau et al., 2013; Eisaman et al., 2018; Zhao et al., 2020; La Plante et al., 2021; Oloye et al., 2021).
Direct CO2 Removal
A range of approaches have been proposed to extract CO2 from seawater, analogous to methods that remove it directly from the atmosphere (Willauer et al., 2011; Eisaman, 2020). Acid approaches exploit acidic conditions created around the anode to shift the equilibrium of the carbonate system (see Box 8.1) toward a greater concentration of aqueous CO2, which is evolved/degassed from the solution and collected for permanent storage (de Lannoy et al., 2012; Datta et al., 2013). Basic approaches exploit high-pH conditions created around the cathode to shift the equilibrium of the carbonate system toward a greater concentration of bicarbonate and/or carbonate ions (La Plante et al., 2021). This creates conditions in which carbonate precipitation can occur and promotes an increase in aqueous CO2, which may be evolved and collected similarly to the acid approach (Rau, 2008; de Lannoy et al., 2012). Dissolved inorganic carbon is removed and collected as both solid carbonate residues and as evolved CO2 gas. Basic approaches that force the precipitation of solid carbonate without restoring alkalinity do not result in the net removal of CO2 from the atmosphere, although they reduce the concentration of CO2 that is dissolved in solution. In both approaches, the base and acid streams are recombined and discharged back into the ocean.
Alkalinity Creation
It is possible to use electrochemical processes to increase the alkalinity of seawater, and/or to force the precipitation of solid alkaline materials (i.e., hydroxide minerals). In the former process, reactions at the cathode increase the alkalinity of the surrounding solution, which can then be discharged into the ocean. In the latter, the reaction cell is separated by an ion-selective membrane, a physical barrier, or internal hydrodynamics, and seawater or brine is introduced into the cathode compartment (Rau et al., 2013; Zhao et al., 2020; La Plante et al., 2021; Oloye et al., 2021). The potential difference across the cell promotes the migration of ions (usually sodium) across the barrier/cell, and ultimately promotes the formation of solid metal hydroxide residues (e.g., brucite or portlandite) that could be added to the ocean to increase its alkalinity.
These two broad categories of ocean CDR electrochemical engineering are, in some cases, not mutually exclusive, and could be deployed as a hybrid approach that both extracts CO2 from seawater, in the form of a gas, or as mineral carbonates, while increasing the alkalinity of the effluent solutions. In this chapter we do not refer to applications for direct CO2 reduction (e.g., to produce syngas, fuels, or chemicals), but specifically focus on approaches for removing or absorbing CO2 from/into seawater.
According to criteria described in Chapter 1, the committee’s assessment of the potential for electrochemical processes, as an approach to CDR, is discussed in Sections 8.2–8.5 and summarized in Section 8.6. The research needed to fill gaps in understanding how electrochemical processes can be used as a mechanism for durably removing atmospheric CO2 is discussed and summarized in Section 8.7.
8.2 KNOWLEDGE BASE
Electrochemical Engineering
The relationship between electricity and chemistry has been explored since the 18th century, with anatomists demonstrating movement of muscles with the application of electricity (Galvani, 1841). This was followed by a spate of new discoveries in the early 19th century including the production of hydrogen and oxygen through electrolysis (Grove, 1839), electroplating, and a chemical-based description of a galvanic cell. The first fuel cell was demonstrated in the 1830s (Grove, 1839), and the Hall-Heroult process for producing aluminum metal from bauxite ore was invented in the 1880s (Charles, 1889).
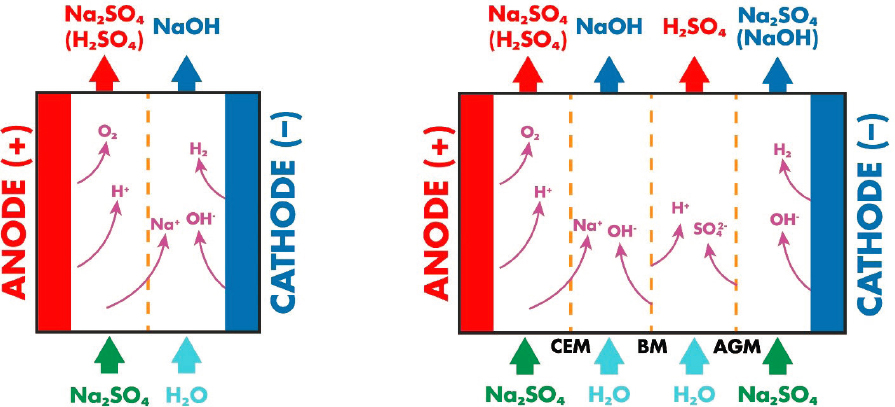
In processes that use electricity to drive reactions, a potential difference is applied to a liquid using electrodes. Positively charged cations in the solution migrate to the cathode (negatively charged electrode), which promotes reduction reactions, and the negatively charged anions migrate to the anode (positively charged electrode), which promotes oxidation. Typically, these reactions are undertaken in a “cell,” and the electrodes are separated by a permeable separator. Figure 8.1 shows a simplified schematic of a diaphragm cell used in the production of chlorine and sodium hydroxide, chlor-alkali.
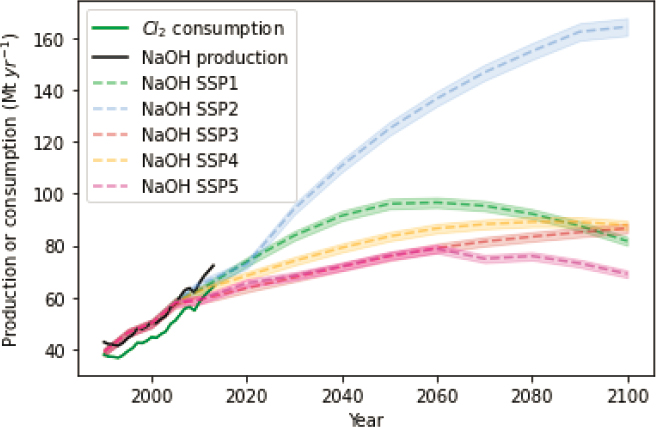
The two largest industrial users of electrochemistry are in the production of aluminum, in which 50 million tonnes (Mt) are produced annually (USGS, 2021) and chlor-alkali (sodium hydroxide and chlorine), in which 72 and 65 Mt are produced annually, respectively (Egenhofer et al., 2014; Renforth, 2019). Innovation in these industries over the 20th century was largely concerned with improving energy efficiencies, reducing cell costs, and improving the purity of the final product.
Design Choices in Electrochemical Engineering
A range of design choices may be available in the construction and the operation of electrochemical reaction systems. These choices are made so that overpotentials at the electrodes or within the cell are minimized, while maximizing current and yield efficiency either when using electricity as an input, or in the case of multiple energy inputs such as electrons and photons/phonons (Nørskov et al., 2004). An exhaustive discussion of these technology options for ocean-based CDR is beyond the scope of this report, but a few possibilities are articulated below (Brinkmann et al., 2014; Paidar et al., 2016; Yan et al., 2020).
Choice of Electrolyte
The electrolyte serves the dual purpose of conducting electric current between electrodes and as a reactant in the chemical processes. The ohmic overpotential (ηohmic, mV) created by the elec-

trolyte is inversely proportional to its conductivity (equation 8.6; Uhlig and Revie, 1985; O’Brien et al., 2005):
| (8.6) |
where, k is the specific conductivity (µS/cm), d is the spacing between electrodes (cm), and i is the current density (A/m2). A less conductive electrolyte will result in greater resistive losses for a given geometry and current density of an electrolysis cell. The composition of the electrolyte also influences resistive losses resulting from mass transport of ions across boundary layers at the electrodes and the cell divider, such that a limiting current (ilim) is imposed on the system (equation 8.7; O’Brien et al., 2005):
| (8.7) |
where, [X] denotes the concentration of either the cation or anion in bulk solution (mol/kg), δ is the diffusion layer thickness (m), and Dx is the diffusion coefficient of the anion or cation X (m2/s). Finally, the reaction of an electrolyte may create unwanted reaction products (e.g., hypochlorite ions in the chlor-alkali process) or reverse reactions may occur (e.g., the production of water in alkaline electrolysis). These undesirable reactions consume some of the electrical energy of the process, resulting in an inefficiency (Bennett, 1980). The ratio of the theoretical electric current needed for creating the desired product to the total electric current used by the system is the Faraday or current efficiency (Bard and Faulkner, 2001). With appropriate process engineering, current efficiencies of >95 percent have been reported in the chlor-alkali industry (Brinkmann et al., 2014).
Table 8.1 shows the possible ohmic overpotential and limiting current density for a range of mineral-saturated electrolytes. For instance, use of an NaCl-saturated brine, a common feedstock in chlor-alkali, results in low ohmic overpotential (<40 mV) and high limiting current densities
(>100 kA/m2). While it may be desirable to exclude chlorine (Cl) from the electrolyte for the CO2 removal (CDR) processes described below, an electrolyte based only on calcium (Ca)/magnesium (Mg) carbonates or hydroxides would result in high ohmic overpotential and low current densities. For comparison, current densities of 1–6 kA/m2 are typical in chlor-alkali (Brinkmann et al., 2014).
Choice of Cell Divider
Early electrolysis experiments used electrolysis cells in which the cathode or anode were not separated (undivided cells; Konishi et al., 1983), as is commercial best practice for aluminum production (Haupin, 1983) or electrochemical wastewater treatment (Chen, 2004). However, to increase selectivity, limit unwanted reactions, and increase product purity (Paidar et al., 2016), the two halves of the electrolysis cell are often separated (or “divided”) by either a diaphragm or an ion-permeable membrane, the former allowing both ion and solvent transport, while the latter allows only ions. Diaphragm materials that are porous, thin, and chemically stable in the electrolyte and cell operating conditions are preferred, with asbestos being used extensively in the past, and polymer membranes finding use in more recent times (Strathmann, 2004).
Choice of Electrode Material
Electrode materials (i.e., herein referring predominantly to the active/electrocatalyst surface, and not the current collector) have been developed and used to improve the performance and to reduce the cost of electrolysis cells. The lowest exchange current densities and the highest catalytic properties for the lowest material cost are desirable. Nickel-based alloys are used extensively in alkaline water electrolysis (for both electrodes) and for the cathode in chlor-alkali (the anode is often composed of ruthenium-titanium oxides; Brinkmann et al., 2014). Challenges associated with disposal of chlorine gas created during the electrolysis of seawater have promoted the development of oxygen-selective electrodes (e.g., Izumiya et al., 1997; Petrykin et al., 2010; Vos et al., 2018, 2019; Okada et al., 2020). Chlorine evolution at the anode involves fewer electrons and intermediary products compared with oxygen, and thus has a kinetic advantage such that chlorine is typically the only detected gas (Vos and Koper, 2018). Reduction in chlorine production could be essential in the development of some of the systems considered below, although oxygen evolution could accelerate electrode degradation (Kolotyrkin et al., 1988) and will also need to be carefully managed in systems that also evolve hydrogen.
Choice of Cell Configuration
In electrodialysis the electrodes are separated by an ion-selective membrane, with the intention of separating the acid and base streams (Figure 8.2). It is possible to separate the electrodes with additional membranes that are selective for either anions or cations (bipolar membrane electrodialysis). A bipolar membrane configuration is intended to promote water dissociation into hydrogen ions (H+) and hydroxide (OH−) (rather than ion transport in monopolar membranes) (Pärnamäe et al., 2021), producing separate streams of acid and base.
Description of Proposed Systems
A range of systems have been proposed for CDR or ocean alkalinity enhancement (OAE), summarized in Table 8.2. For simplicity we represent the products of electrolysis as acid and base. Both CDR processes “‘swing” alkalinity of seawater to extract the largest amount of dissolved inorganic
carbon—whether as a gas or as mineral carbonate. However, there is no net impact on alkalinity when acid and base streams are recombined.
Several systems have been proposed that do not use electrochemistry, but either remove CO2 from seawater or increase its alkalinity. Willauer et al. (2009) considers a system that filters bicarbonate, and Willauer et al. (2011) investigated CO2 extraction using ion exchange resins. These experiments were conducted on solutions with elevated partial pressure of carbon dioxide (pCO2). Given that they do not modify alkalinity, the water requirements at ambient CO2 pressures may be prohibitive (see discussion in Section 8.3). Davies (2015) investigated the thermal decomposition of desalination rejected brine. Similar to seawater electrolysis, the products are solid magnesium hydroxide (Mg[OH]2) and liquid hydrochloric acid (HCl), although the thermal energy requirements possibly limit wide application
Acid Process
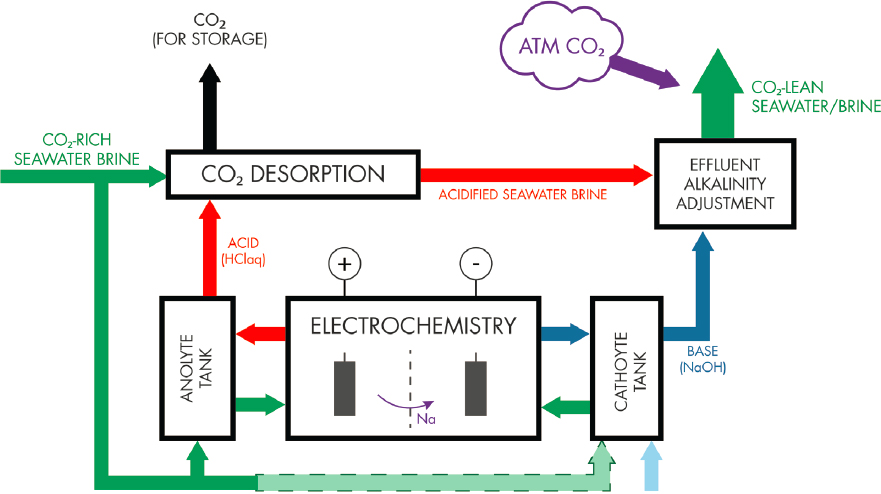
CO2 Removal
This approach (Figure 8.3) uses electrolysis to create streams of acid (HCl, at the anode) and base (NaOH, at the cathode). When the acidic anolyte is mixed with CO2-“rich” seawater or brine, the acidification shifts the equilibrium of the carbonate system toward CO2, which is vented and can be subsequently pressurized and geologically stored. The acidified CO2-lean seawater is mixed with the base liquid stream and then released to the ocean where it absorbs CO2 from the atmosphere to an extent described by its alkalinity and temperature.
In the system explored by Eisaman et al. (2012) and de Lannoy et al. (2018), a small proportion of influent seawater is treated or demineralized using nanofiltration, mineral precipitation, CO2 desorption, and resin towers (e.g., as typical of a system used in the chlor-alkali process to limit electrode fouling) before being passed into a three-compartment bipolar membrane electrodialysis system. The acid generated in electrolysis (0.4–2 mol/kg) is used to acidify 160–800 times the volume of seawater. Prior to acidification, nitrogen (N2) and oxygen (O2) are extracted at 30-mbar pressure, such that pure CO2 gas is extracted at 30–80 mbar pressure (i.e., by vacuum stripping).
TABLE 8.1 Comparison of Electrochemical Properties of a Range of Mineral-Based Electrolytes
| Electrolyte (equilibrium mineral phase) | Saturated Cation Conc. (log10 mol/kg) | Saturated Anion Conc. (log10 mol/kg) | Anion | Ionic Strength (mol/kg) | Conductivity (µS/cm) | Electrolyte Ohmic Overpotentiala (mV) | Mass Transfer Limit Current Density—Cationb (kA/m2) | Mass Transfer Limit Current Density—Anionb (kA/m2) |
|---|---|---|---|---|---|---|---|---|
| NaOHc | 0.80 | 0.69 | OH− | 4.86 | 319,321 | 6 | 81 | 246 |
| KOHc | 0.65 | 0.54 | OH− | 3.50 | 230,963 | 8 | 85 | 177 |
| Ca(OH)2 (portlandite) | −1.82 | −1.56 | OH− | 0.04 | 2,731 | 659 | 0.2 | 1.4 |
| Mg(OH)2 (brucite) | −4.10 | −3.80 | OH− | 0.0002 | 17 | >100,000 | <0.01 | <0.01 |
| NaCl (halite) | 1.12 | 1.12 | Cl− | 6.88 | 450,675 | 4 | 170 | 261 |
| CaCl2 (antarcticite) | 0.93 | 1.23 | Cl− | 8.38 | 548,029 | 3 | 129 | 664 |
| MgCl2 (bischofite) | 0.93 | 1.23 | Cl− | 11.85 | 773,002 | 2 | 116 | 664 |
| Na2CO3 (Na2CO3) | 1.00 | 0.70 | NaCO3− | 7.25 | 474,900 | 4 | 127 | 53 |
| K2CO3 (K2CO3•1.5H2O) | 1.10 | 0.80 | CO32− | 18.85 | 1,224,502 | 1 | 237 | 111 |
| CaCO3 (calcite) | −3.92 | −4.09 | HCO3− | 0.0004 | 26 | >50,000 | <0.01 | <0.01 |
| MgCO3 (magnesite) | −3.74 | −3.95 | HCO3− | 0.0006 | 41 | >40,000 | <0.01 | <0.01 |
| Na2SO4 (thenardite) | 0.68 | 0.38 | SO42− | 3.84 | 253,241 | 7 | 61 | 49 |
| CaSO4 (gypsum) | −1.80 | 0.20 | SO42− | 0.05 | 3,127 | 576 | 0.2 | 32 |
| MgSO4 (epsomite) | 0.87 | 0.87 | SO42– | 3.38 | 223,119 | 8 | 101 | 150 |
| Seawater | – | – | 0.64 | 43,119 | 42 | – | – |
NOTES: Solution saturation concentrations were calculated using PHREEQC v3 (Parkhust and Appelo, 1999) and the LLNL.dat database. Mineral phases (in parenthesis) were brought into equilibrium with deionized water at 25°C and 1 bar.
a Assuming - 5mm electrode spacing and a current density of 6 kA/m2 across 3-m2 cell divider.
b Assuming a diffusion layer thickness of 0.01 mm.
c Considers a solution of 25% w/w.
TABLE 8.2 Summary of Ocean-Based Electrochemical Approaches for CDR from the Atmosphere
| Direct CDR—acid process | The acid stream from the anode in the electrochemical cell is used to decrease the pH of seawater to evolve CO2. The base stream from the cathode is then mixed with the decarbonized seawater to capture additional CO2 from the air, resulting in a continuous closed cycle where CO2 is effectively removed from the air via seawater. |
| Direct CDR—base process | The base stream from the cathode in the electrochemical cell is used to precipitate carbonate from seawater with or without the evolution of CO2. |
| Ocean alkalinity enhancement (OAE)—seawater/brine electrolysis | Using seawater as the electrolyte, sodium hydroxide (NaOH) is concentrated at the cathode (which is added to the ocean to increase alkalinity; see Chapter 7). The acid stream from the anode is neutralized through reaction with silicate rocks. |
| OAE—water electrolysis | Similar to seawater/brine electrolysis but using an alternative non-NaCl-based electrolyte (e.g., based on Ca or Mg). |
| OAE—salt recirculation | Similar to seawater/brine electrolysis but the salt form, through reaction with silicates, is recycled back into the catholyte. |
| Hybrid approaches | A combination of electrochemical approaches that results in an increase in ocean alkalinity and the removal of CO2 from seawater; e.g., as a gas, or in mineral carbonates. |
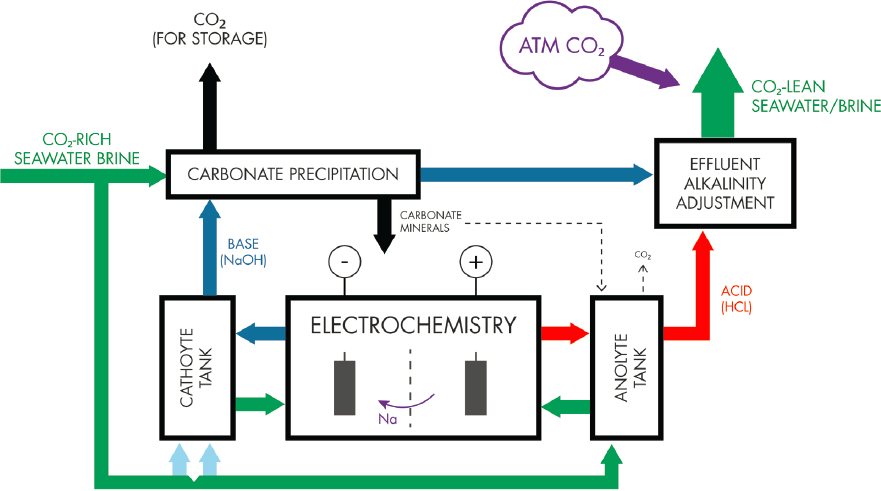
Base Process
In this approach the base liquid produced by electrochemistry is mixed with seawater/brine to force the precipitation of carbonate minerals. Like the acid process, carbonate precipitation shifts the equilibrium of the carbonate system toward CO2, which if not suitably compensated (e.g., by addition of base) would require venting, pressurization, and storage. The basic seawater is mixed with the acid stream from the electrodialysis unit before being returned to the ocean. Addition of base to seawater increases pH and potentially results in the oversaturation of a range of minerals. Relatively smaller additions of NaOH would oversaturate carbonate minerals, leading to their precipitation. But, if the addition rate of NaOH was greater than the carbonate precipitation rate, then it is possible that a higher pH could also lead to the precipitation of calcium or magnesium hydroxide minerals, which would result in a net reduction in alkalinity of seawater if they were not redissolved (Figure 8.4). De Lannoy et al. (2018) suggest that maintaining a pH between 9.3 and 9.6 would prevent this. Figure 8.5 shows the effect of the progressive addition of NaOH to seawater at different rates (slow and fast) and the resulting saturation indices for brucite (Mg(OH)2), a hydrated magnesium carbonate mineral hydromagnesite (Mg5(CO3)4(OH)2•4H2O), and strontianite (SrCO3) for solutions equilibrated with calcite (Figure 8.5a, i.e., by slow addition of NaOH to allow calcite to precipitate) and those unequilibrated to a mineral phase (Figure 8.5b, i.e., by rapid addition of NaOH).
Production of Alkalinity by Seawater Electrolysis
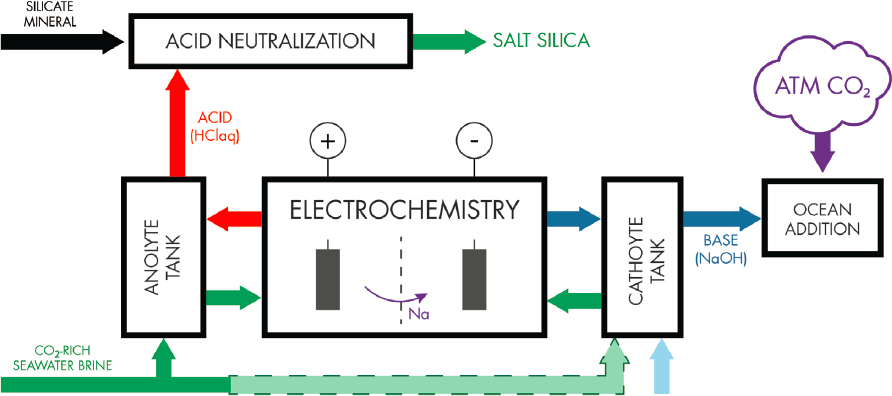
A system similar to that used in the chlor-alkali process could also be used for the production of solid or liquid bases for ocean alkalinity enhancement (Chapter 7). In these systems, seawater or brine are introduced into the anolyte/anode. A potential difference between cathode and anode encourages the migration of sodium across the cell divider such that an acid solution is produced at the anode and a basic solution at the cathode. In the system espoused by House et al. (2007) chlorine gas produced

at the anode and hydrogen gas at the cathode are combined in a fuel cell to produce hydrochloric acid. The acid is then neutralized through the reaction with a silicate mineral to produce a salt and silica (equation 8.8), which will need to be disposed of, or could potentially be valorized in other industries. The base is discharged into the ocean (as a solid or a liquid) to increase ocean alkalinity (Figure 8.6). Davies et al. (2018) presents a system that is coupled to the reject brines from reverse osmosis, where the alkalinity is removed as a solid MgOH2 base. This system is simulated by Tan et al. (2019), who suggest that reverse osmosis water production could be carbon negative, providing an appropriate carbon incentive ($10–$100/t CO2) and/or revenue stream from HCl production if implemented and low-carbon electricity is used.
| 4HCl(aq) +MMg2SiO4(s)→2MgCl2(s/aq)++SSiO2(s) +HH2O(l) | (8.8) |
Production of Alkalinity by Water Electrolysis
A simplified version of a system proposed by Rau (2008) and Rau et al. (2013) is shown in Figure 8.7. In this system a basic or ultrabasic silicate mineral is reacted directly with the anolyte to produce a solution rich in cations (e.g., Ca and Mg), silicon, and other elements in the rock. This solution is passed to the anode chamber of an electrolysis cell, and the cations selectively migrate to the cathode across the cell divider. The base concentrated solution can be passed to a system for concentration and precipitation of Ca or MgOH2 for subsequent addition to the ocean, or the elevated alkalinity solution could be added directly to the ocean. A solution containing only Ca2+ or Mg2+ charge balanced by OH− ions will likely result in prohibitive ohmic losses in the electrolyte or mass transport losses at the electrode (Table 8.1). As such, the presence of a highly soluble salt (e.g., SO42− used by Rau et al., 2013) is essential.
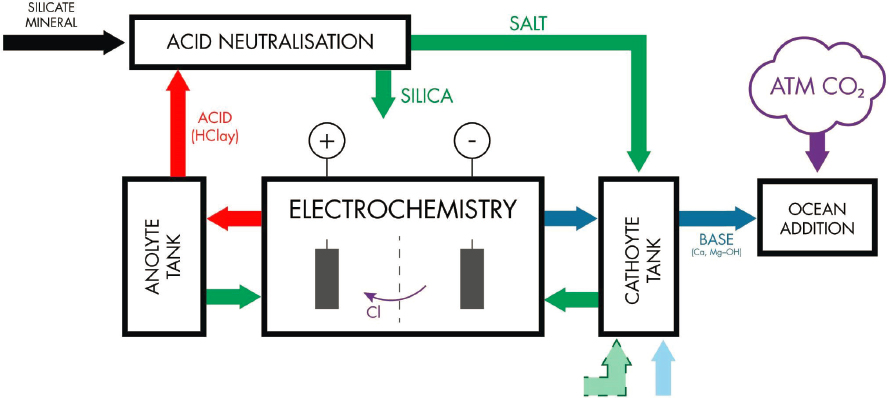
Production of Alkalinity by Salt Recirculation
This involves a system that promotes the migration of anions (e.g., Cl−, SO42−) across the electrolysis cell divider to form an acidic solution for neutralization through reaction with basic or ultrabasic (Ca, Mg) silicate minerals (hypothesized by Rau et al., 2013). The solid or aqueous

salt is then added to the catholyte, resulting in an accumulation of Ca or Mg, and an increase in alkalinity (Figure 8.8).
8.3 EFFICACY
Achievable Scale
When considering electrochemical methods for ocean-based carbon removal, it is important to contextualize the scale that such processes could achieve if they were to be successful. As such, important considerations around achievable scale are based around (see Chapter 1 for discussion on scale:
- The need/consumable demand for stoichiometric additives (e.g., acids, bases, salts),
- The materials of construction of electrochemical systems (e.g., alloys),
- The net energy intensity of the life cycle of the process and its supply chain, and
- The grid emissions factor of electricity that is used to power the approach.
In brief, first, unless reagents and additives are effectively perfectly reusable (i.e., the process features a vanishingly small consumable additive demand), independent of their cost, on account of their global production levels (e.g., NaOH is produced using the chlor-alkali process at a level of around 70 Mt/yr (World Chlorine Council, 2017)), and the energy intensity of their production (e.g., for NaOH, 2.5 MWh/t (Chlistunoff, 2005)), the use of or need for stoichiometric additives provides a means to estimate the practical viability of a CDR process. In this context, electrochemical methods have a particular advantage because they may be capable of producing acidity/basicity, in situ, without a need for ancillary additives, synthetic electrolytes, etc.; in effect, the additive demand can be fulfilled by, ideally, zero-carbon electrons. Second, it is critical that the materials of construction, particularly the electrode materials and reactors are abundant. These may include, but are not limited to, polymers, alloys, and or carbon composites, which feature a basis for widespread production today.
Finally, since natural gas may continue to be a significant source of energy (electricity), generally in the short to medium term, it would be attractive if carbon removal processes featured a
net (i.e., uncompensated by the potential embodied energy benefit of any co-produced hydrogen) energy intensity/demand of less than 2.4 MWh/t CO2 removed—to deliver a (net) negative carbon removal outcome.1 Maximum (net) energy intensity is quantified by assessing the specific intensity ratio (SIR) of the energy source, that is, the reciprocal of the grid emissions factor (e.g., for natural gas combustion–based electricity generation, this is around 0.42 t CO2 emitted per MWh of electricity produced;2 see Section 8.3). Of course, it is necessary that, in general, the net energy intensity be substantially inferior to the maximum value to realize the highest CDR benefit for example, as ensured by use of renewable energy as far as possible, particularly during periods of excess generation capacity (i.e., when curtailment is ongoing). All that said, it is particularly desirable to develop electrochemical processes that have the potential to produce valuable co-products, for example, hydrogen, oxygen, and/or minerals (see Section 8.4). The former are particularly attractive because if these “clean fuel” co-products can partially power the electrochemical process, they in effect allow for reduced reliance on fossil fuel sources (e.g., up to 50 percent less fossil-fuel energy would be required depending on the co-produced hydrogen yield). Also, in an energy storage–constrained market, they offer a means to bridge the energy gap between time periods when renewable electricity may be in short supply (e.g., at night) while reducing the net carbon intensity of the fuel source that is used to power the CDR process (e.g., when natural gas–based electricity may be a possible energy input).
Additionality and Downstream Effects
Despite their enormous potential, electrochemical processes that couple with the world’s oceans may exert unintended consequences. Such consequences include some that we can imagine today, and other nonlinear and downstream effects that may only be ascertained when electrochemical CDR processes are operated at the pilot or larger scales. In addition to general issues associated with ocean-based CDR, electrochemical CDR processes seek to (1) remove dissolved CO2 from seawater as a means of reducing its acidity or (2) alkalinize seawater as a means of enhancing its
___________________
1 Here the term “removal” implies end-to-end carbon abatement including capture, sequestration, and immobilization for periods generally well in excess of 10,000 years.
2 See https://www.eia.gov.
capacity to absorb and store additional CO2 from the atmosphere in the form of bicarbonate species. Such acidification or alkalinization of seawater will, deliberately, affect the water chemistry (e.g., pH, distributions and concentrations of species such as CO2 and bicarbonate in solution and within mineral precipitates, turbidity, etc.). Such changes in water chemistry may affect ocean ecology and marine organisms (e.g., by changing the optimal pH for growth and reproduction) and should therefore be limited as much as possible.
Permanence
Electrochemical methods for ocean-based CDR seek to (1) extract dissolved CO2 in the form of a gas, after which it will be compressed and sequestered in geological formations; (2) stabilize dissolved CO2 in the form of aqueous bicarbonate (HCO3−) species, for example, when alkalinity enhancement of the aqueous phase allows for the additional drawdown of atmospheric CO2; and/or (3) immobilize CO2 in mineral carbonates. If effectively implemented, particularly the latter approaches (2 and 3), can ensure the durable and effectively permanent sequestration of CO2 at Earth’s surface while eliminating the risk of accidental release (La Plante et al., 2021) due to, for example, fault activation in geological sequestration (Jahediesfanjani et al., 2018). Notwithstanding changes in sea-surface temperature, CO2 stabilized in the form of bicarbonate anions is expected to remain stable in surface waters (seawater) for periods on the order of 100,000 years (Falkowski et al., 2000; Caldeira et al., 2018). CO2 stabilized within mineral carbonates, as highlighted by the persistence of limestone deposits on Earth’s surface, is expected to remain stable for periods on the order of hundreds of millions of years (Lackner, 1995).
Monitoring and Verification
Electrochemical methods, on account of being contained engineering processes, are readily amenable to assessments of mass, energy, and CO2 balances, that is, both embodied in and as related to CDR, within the boundaries of the process. For example, processes that generate acidity or basicity can be readily instrumented to assess the extent of acid and base generation within a liquid stream (e.g., of the anolyte and catholyte), the amount of hydrogen, oxygen, and/or chlorine produced (e.g., in the case of seawater electrolysis) or CO2 evolved (in the case of CO2 stripping processes), the amount of solid precipitates formed (e.g., mineral carbonates and/or hydroxides) or minerals dissolved (e.g., silicate rock dissolution), and the consequent energy expenditures and seawater fluxes. Such monitoring can be carried out by analysis of the influent and effluent compositions, typically within electrochemical cells or reactors, and of the gases evolved within a closed headspace. The challenge with electrochemical processes, however, is that their CDR basis may often be indirect. For example, except in the case of CO2 stripping or solid carbonate mineral formation, these processes often rely on the alkalinization of seawater directly or by the dissolution of minerals within it (e.g., brucite, silicate rock, etc.) to enhance its CO2 storage capacity. The enhancement of the CO2 storage capacity results in the additional dissolution of atmospheric CO2 in or its absorption into seawater, which in turn results in CDR. If mineral dissolution may occur within the system boundary, the temporal dynamics and the amount of atmospheric CO2 removal that will occur can be readily established. But, if mineral dissolution may occur slowly, and beyond the process boundaries (e.g., following discharge of the effluent into the ocean), it will be necessary to rely on an indirect basis of quantifying the CDR benefit.
8.4 SCALABILITY
Resource Availability
A large, and practically unlimited, elemental reservoir is found in the ocean for the production of acid and base creation (HCl and NaOH). If all NaCl in seawater were split, the equivalent of 26 × 106 Gt of NaOH would be produced, which could capture 30 ×106 Gt of CO2 (equivalent to 1,000x more than the carbon in the Earth system).3 Furthermore, terrestrial brines (e.g., produced water, desalination brines) could also provide resources for such acid and base production, although to a smaller extent. From stoichiometry and charge balance considerations, 2 mol of base (NaOH) are needed to neutralize 1 mol of CO2, or more than 1 mol of acid is needed to dissolve 1 mol of alkaline mineral (e.g., CaCO3, MgSiO3, etc.). So, to effect 500 Mt/yr of CDR, an equivalent quantity of acid and base is needed. This is far larger than current scales of production ~70 Mt/yr for NaOH (and most future projections, see Figure 8.9) and ~300 Mt/yr for H2SO4.4 Mined salt is a common feedstock for chlor-alkali, but as such, this requires not only the development of new facilities for acid and base production but also new infrastructure to store, transport, or dispose of acid and base materials.
The ocean offers a considerable resource for absorbing gaseous CO2. Systems designed to extract CO2 from seawater unavoidably create mass handling challenges. The ocean contains approximately 2 mmol/kg (88 g CO2/m3) of dissolved inorganic carbon; aqueous CO2 constitutes only a small proportion of this (~10 µmol/kg at atmospheric pressure). The majority of the carbon is contained in the form of HCO3- and CO32− ions. As such, without modifying alkalinity, it may only be possible to extract hundreds of milligrams of CO2 per cubic meter of seawater. While the prevalent volumetric concentration of CO2 in the ocean (>80 g CO2/m3) is greater than that in the atmosphere (<1 g CO2/m3), the mass concentration is less (0.09 and 0.6 gCO2/kg in the ocean and atmosphere, respectively). A facility designed to remove 0.1 Mt CO2/yr from seawater would need to treat 1.3 Gt seawater/yr, or about 3 million m3/day, which is 5–10 times greater than the world’s largest desalination facilities (Eke et al., 2020). In other words, treating a water flux as great as present-day global desalination would amount to around 1–2 Mt CO2/yr removal. Treating a similar seawater volume to increase its alkalinity could remove ~90 Mt CO2/yr. Thus, there may be advantages to processes that create a concentrated solid or liquid base, ex situ or in situ, and distribute this in the ocean.
Some of the processes described above require the extraction, processing, and dissolution of rock either to increase alkalinity or to replenish alkalinity, which is lost during carbonate precipitation. The scalability implications of rock extraction is discussed in Chapter 7. If CO2 is extracted from seawater, there is a need to store this fluid-state CO2, for example, either in terrestrial reservoirs or by pumping it into the very deep ocean.5 It is likely that large volumes of CO2 derived from either the atmosphere or captured emissions will need to be disposed of over the next century. This challenge was explored in a recent National Academies report (NASEM, 2019, Chapters 6 and 7) and elsewhere (Kelemen et al., 2019).
___________________
3 Assumes the sodium concentration of seawater is 0.4 mol/L (Pilson, 1998) and the volume of the ocean is 1.3 × 109 km3 (https://ngdc.noaa.gov/mgg/global/etopo1_ocean_volumes.html). The Earth system contains 871, 1,400, 450, 900, 700, 1,750, 37,100 (~43,000 in total) Gt C in the atmosphere, permafrost, soils, surface ocean, dissolved organic carbon in the ocean, shallow sediments, and deep ocean, respectively (Canadell et al., 2021).
4 See https://www.essentialchemicalindustry.org/chemicals/sulfuric-acid.html.
5 Note that the liquid CO2 relevant to a deep-ocean disposal approach relies on the basis that liquid CO2 is denser than liquid water.
Low-Carbon Electricity
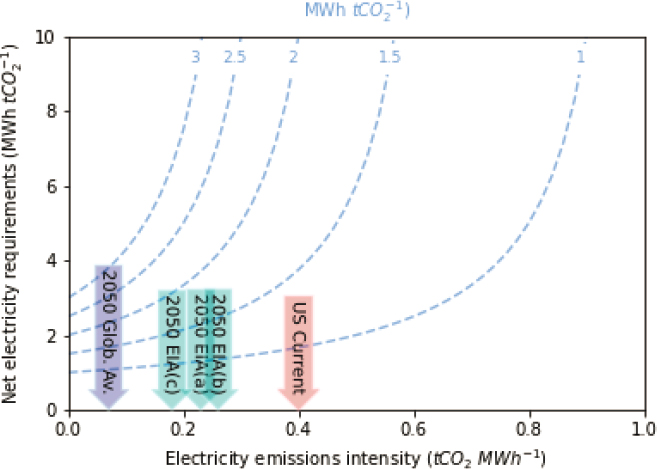
Global annual electricity supply is currently 25,000 TWh and will possibly increase to ~35,000 by 2040 (EIA, 2021). If an electrochemical approach consumed ~2 MWh/t CO2 removed (see below), then 1 Gt CO2/yr would require around 2,000 TWh/yr (20 percent addition on the projected increase). The approaches described above will likely require low- or zero-carbon electricity to be feasible. While the supply and generation capacity of renewable electricity are increasing, current forecasts estimate that fossil fuels will contribute ~45 percent of electricity generation for the next three decades at least (EIA, 2021). As such, it is necessary to reduce the energy intensity of CDR approaches to below 1.5–9.5 MWh/t CO2 (i.e., an inverse of the 105–680 kg CO2/MWh grid emissions intensity of electricity produced in the United States;6Figure 8.10).
It may be possible to use excess electricity generation at times of the day in which demands of the grid are lowest, and thus the power is “cheap.” Such electricity is only available for no more than ~8 hours when curtailment is in effect.7 To operate a capital-intensive facility using such electricity implies accepting a capacity factor of 30 percent, which produces a capital inefficiency (due to lack of operation for the majority of the day), and a need to oversize the plant to meet production goals. Although such reduced utilization could be remedied, it requires access to cheap long-duration electricity storage and/or net emission technologies to produce their own fuels (e.g., hydrogen) to power operations when curtailment is not in effect or cheap renewable electricity is not available.
Geospatial and Adjacent Infrastructure Considerations
Unquestionably the need for seawater in electrochemical ocean CDR requires the siting of such processes and plants at sites that offer ready access to seawater. While the optimal attributes of sit-
___________________
6 See https://www.epa.gov/egrid/data-explorer.
7 See https://www.pge.com.
ing, for example, in the open ocean, along the coast, or in river deltas are not fully clear, there are adjacent infrastructure considerations that are relevant: (1) seawater access, (2) low-carbon energy supply, (3) infrastructure for supply of raw materials and disposal of waste, and (4) labor.
First, a significant cost associated with desalination plants (and other facilities that require seawater) is the infrastructure for seawater intake. Thus, colocation at sites that host water intake infrastructure (e.g., desalination plants, power plants, etc.) is valuable not only for reducing the overall capital costs of eventual commercial plants, but also conceivably for the potential to utilize operating water intake permits of existing facilities (see section on Governance below). Second, the most effective means for CDR implies the use of renewable energy. Thus, it is important to locate eventual facilities in regions that feature substantial potential for renewable energy generation including wind and solar (either photovoltaic or concentrated solar power based). Third, if CDR is accomplished in a manner that yields a fluid, or solid, it is necessary to build out infrastructure including pipelines (and compression stations) to convey the CO2 to sequestration sites, or to develop solid waste handling and valorization facilities (e.g., for the potential use of mineral carbonates and hydroxides as construction materials). Although some of these facilities already exist (e.g., landfills for solid waste handling), given the enormous scale of CDR required, it would be necessary to expand such facilities considerably, to achieve scale relevance.
8.5 VIABILITY AND BARRIERS
Environmental Impact
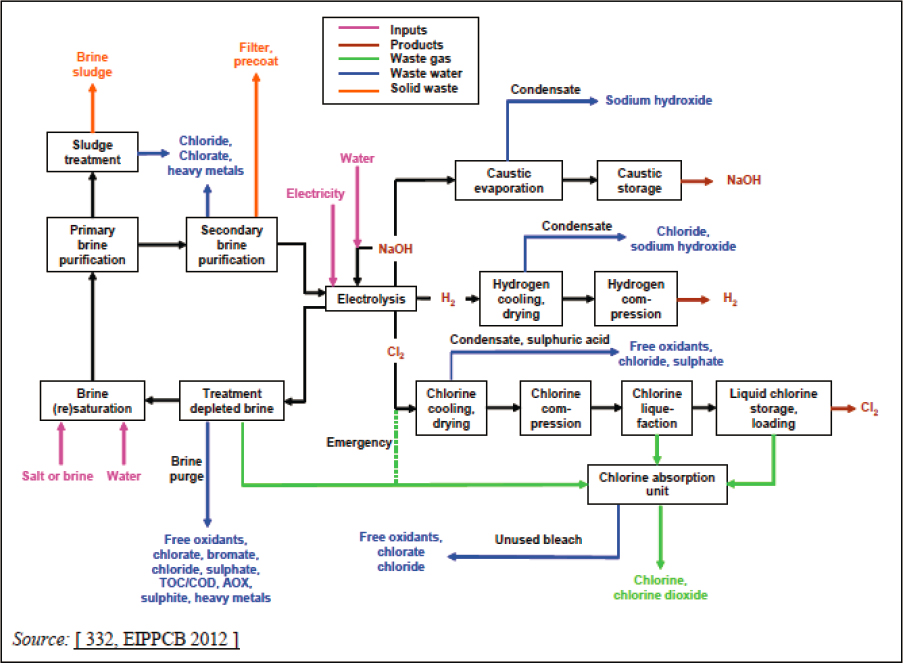
A comprehensive life-cycle assessment for electrochemical ocean-based approaches has yet to be completed. The following provides an overview of potential issues, summarized in Figure 8.11, for best available membrane processes. Mercury (Hg) amalgam systems that were historically used for chlor-alkali typically yielded elevated Hg in effluent waters and surrounding sediments, accompanied by other potentially toxic metals (Arribére et al., 2003), and some early diaphragm systems that use asbestos for their cell divider report particle emissions to water and air. Many of these systems are being replaced by membrane or alternative diaphragm systems, such that their environmental impacts would not be applicable to future ocean-based electrochemical approaches. The environmental impact associated with rock extraction is discussed in Chapter 7.
Gaseous Chlorine Production
In seawater or brine electrochemical reactions (equation 8.5) as relevant to the chlor-alkali and conceptually similar processes, around 1 tonne of chlorine gas is produced for sufficient alkalinity to be generated to remove a tonne of CO2. There are approximately 23 Mt of gaseous chlorine in the atmosphere, most of which is derived from human sources (Khalil, 1999), which impacts atmo-
spheric N, OH, and CH4 concentrations (Zhai et al., 2021). Given that the global chlorine market is on the order of 60–70 Mt/yr (Figure 8.9), systems operating at climate-relevant scales will likely require a highly efficient method of managing and disposing of excess chlorine. It may be possible to reduce or eliminate chlorine emissions by using selective electrodes (i.e., that promote the oxygen evolution reaction as opposed to the chlorine evolution reaction), or by reaction of generated Cl2 with H2 to produce HCl and its reaction with cation-rich silicates to produce salt. In addition to chlorine, CO2 is often reported in waste gases.
Wastewater
Table 8.3 presents ranges of emissions to water for best available technology from chlor-alkai processes. Wastewaters are generated in feedstock brine purification and depleted brine treatment, as condensates from product treatment, or bleach from the chlorine gas absorption unit. It is unclear how applicable these are to the CDR or OAE systems considered in this chapter, but an industrial-scale plant will likely produce some wastewater, the generation and treatment of which is not explored in the literature.
Energy Use
The life-cycle impact of the chlor-alkali industry is dominated by energy consumption in electrolysis, which has implications for both the global warming potential impact and the impact on human health (Garcia-Herrero et al., 2017), with membrane-based processes performing favorably.
Reduced Dissolved Inorganic Carbon in Effluent Waters
Processes that remove CO2 from seawater will, by intention, result in very low dissolved inorganic carbon (DIC) concentrations. For instance, a process that removes 90 percent of the DIC while maintaining alkalinity would result in an aqueous CO2 concentration on the order of pmol/kg and a CO2 partial pressure <1 nanoatmosphere. Low DIC levels are possibly detrimental to autotrophic organisms (Hansen et al., 2007) and may be impacted at the point of effluent addition. This water will eventually mix into the surface ocean and promote CO2 drawdown from the atmosphere. It may be possible to design discharge systems (active sparging or passive cascades) to promote gas exchange and limit these effects.
Co-benefits
In all of the approaches outlined above, hydrogen is a likely product of reactions at the cathode. Currently the United States consumes approximately 10 Mt of hydrogen per year, derived largely from steam methane reforming of natural gas or coal gasification, at costs of $1–$2.5/kg H2, with an emission intensity of 40–60 kg CO2/GJ (DOE, 2020). In the system proposed by Rau et al. (2013), 23 kg H2 is produced for every tonne of CO2 removed (or an emission intensity of −300 kg CO2/ GJ, assuming no emissions associated with the process of its energy supply). The baseline shared socioeconomic pathways (Riahi et al., 2017) suggest hydrogen may constitute 2.5 to 4.2 EJ/yr by 2050, rising to possibly tens of exajoules by 2100.
Processes that neutralize excess acid by reaction with crushed silicate rocks or minerals will create a brine composed of dissolved elements from the rock. This brine is likely to contain high concentrations of silica (which is present in some basic silicate rock ~40–50 mass % as SiO2), aluminum (~15 mass % Al2O3), and iron (5–10 mass % iron oxide). Except for nickel and chromium in ultrabasic rocks, other elements are unlikely to be sufficiently concentrated to facilitate recovery.
TABLE 8.3 Emissions to Water from 1 Tonne of Cl2 Production from Best Available Chlor-Alkali Electrochemical Technology
| Water | Up to 2.7 m3 |
| Free chlorine | 0.001–3.8 g |
| Chlorate | 0.9–3,500 g |
| Bromate | 50–300 mg |
| Chloride | 0.6–1,060 kg |
| Sulfate | 0.07–7.4 kg |
| Organic carbon (as total organic carbon) | 2.5–34 g |
| Metals | Cadmium, chromium, copper, iron, nickel, lead, and zinc (derived from impurities in salt or brine) |
SOURCE: Brinkmann et al., 2014. Licensed by Creative Commons CC BY 4.0.
Some processes also promote mineral carbonate formation. Niche high-value markets exist for these precipitated minerals (i.e., fine precipitated silica as a pozzolan in concrete manufacturing, precipitated calcium carbonate as a paint filler). However, use as construction products may be the more scalable destination.
Energy
Electricity consumption in the electrochemical cell dominates the energy balance of all described systems, and much of the reported values are normalized to mass of CO2 removed from the atmosphere, rather than net removal (which would account for CO2 emissions from electricity generation). The carbon intensity of the U.S. electricity grid is between 1.5 and 9.5 MWh/t CO2,8 so a system that consumes 1 MWh/t CO2 removed, may require between 1.1 and 3.0 MWh/net t CO2 removed. Processes that consume more electricity than the carbon intensity of the grid will emit more CO2 than they remove.
Eisaman et al. (2012) suggest that approximately 1.5 MWh are required per ton of CO2 extracted from their acid bipolar membrane electrodialysis process. Their system uses a stack of nine cells operating at a current density of ~0.4–0.5 (mA/cm2)/Lpm to produce 0.1–0.4 liters per minute of CO2 (at standard temperature and pressure) from 3–6 Lpm of seawater. A technoeconomic assessment of an upscaled process (7 kt CO2/yr) suggests an energy requirement of 3.1 MWh for the acid process and 4.4 MWh for the base process per ton of CO2 removed.9
There is no comprehensive technoeconomic assessment for processes that effect CDR and use electrochemistry to increase ocean alkalinity. House et al. (2007) consider electrolysis of seawater and suggest energy requirements between 0.8 and 2.5 MWh/t CO2 removed, which is consistent with Rau (2008), Rau et al. (2013), Davies et al. (2018), and La Plante et al. (2021), who calculate 1.5–2.3 MWh/t CO2 for similar systems.
Cost
Sodium hydroxide prices are sensitive to both energy costs and volatility in chlorine supply and demand. However, a typical price may be on the order of $300–$400 per tonne, typically delivered
___________________
8 See https://www.epa.gov/egrid/data-explorer.
9 Note that the final use or geological storage of the CO2 was not included within the assessment.
as a 50 percent solution (HIS Markit, 2017). Table 8.4 presents a technoeconomic overview of chlor-alkali production. Although simplified, the levelized cost of NaOH is consistent with market prices. It suggests that using NaOH from current chlor-alkali methods may be on the order of $500–$700/t CO2 removed (without revenue from H2 and Cl2 sale) or $450–$600/t CO2 removed (with revenue). This does not account for acid disposal/neutralization, mineral extraction, valorization of precipitated products, seawater monitoring, or the emissions associated with the process or energy supply.
While chlor-alkali systems are not directly comparable to those considered in this chapter, it demonstrates that electrochemical reaction processes are capital and energy intensive. Although it is possible that many of the components will be different (e.g., no, or smaller, gas handling equipment if the sale or processing of Cl2 or H2 is not a priority; a fuel cell for Cl2 or H2 reaction; or less storage if deployment is integrated). However, the largest single capital component is the electrolyzer (i.e., the reaction half cells and electrodes, equating to 30–50 percent of the capital cost), which is essential for all electrochemical reaction systems, although the relative cost of membranes for electrodialysis systems may be greater and electrode material costs lower. The bare module cost (i.e., without other system components, installation costs, owner costs, or contingency) of an aqueous electrolyzer is shown in Figure 8.12. Those costs, incorporated into typical chlor-alkali process costs (Table 8.4) suggest that using a chlor-alkali system to produce NaOH for ocean alkalinity enhancement may cost on the order of $533–$668/t CO2, without including costs associated with acid disposal. If additional revenue was generated from the sale of Cl2 and H2 gas, then the costs for
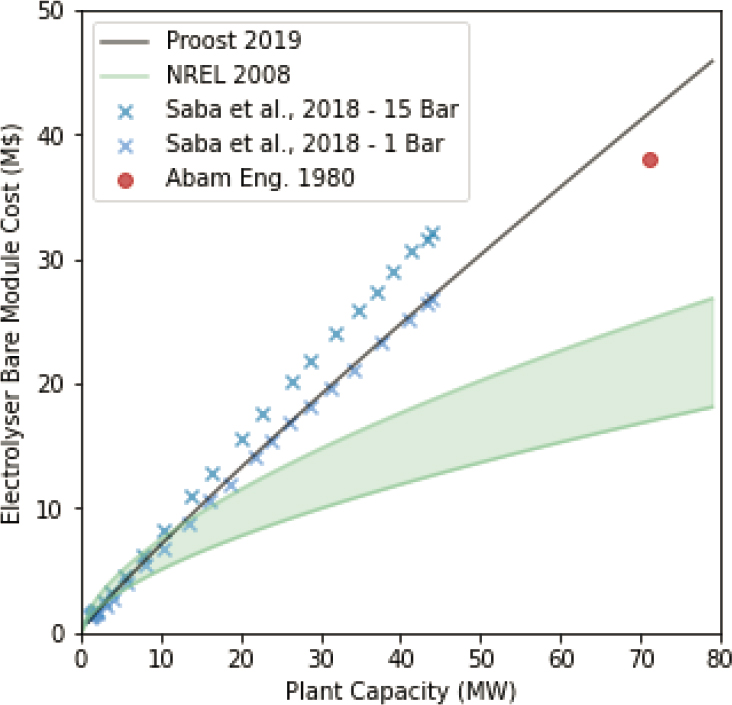
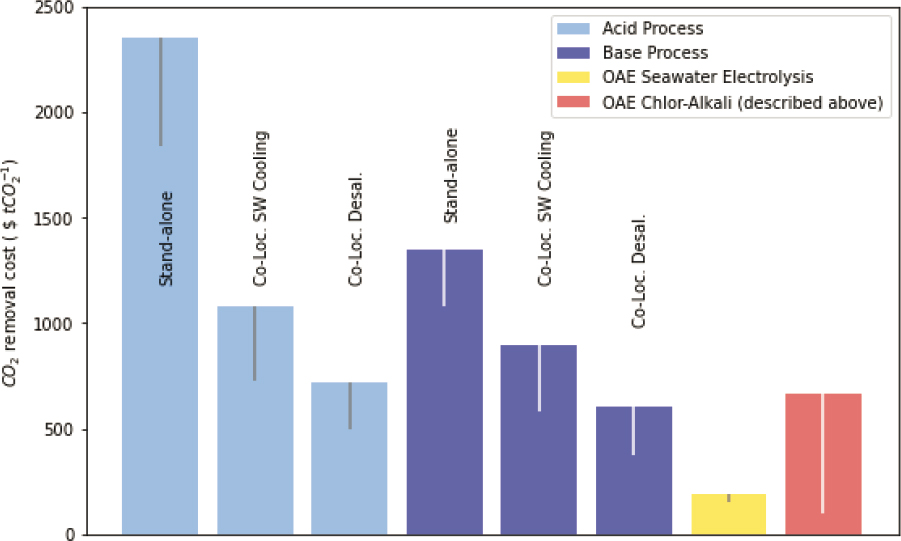
CO2 removal may be 10–15 percent lower. The energy requirements of these systems are approximately 8.8 MWh/t CO2, which is much greater than ~2 MWh/t CO2 estimated by Rau (2008), Rau et al. (2013), and La Plante et al. (2021) (see Figure 8.13). Using this energy requirement together with lower capital requirements (removing rectifiers, boilers, and storage) and reducing raw material costs (e.g., by using seawater as feedstock brine), then the overall cost for OAE by seawater electrolysis could be further reduced to ~$150/t CO2, which is consistent with the preliminary assessments by Rau (2008), Rau et al. (2013), and La Plante et al. (2021). In the absence of cost improvements on conventional electrochemical systems, the cost of ocean alkalinity enhancement through electrochemical methods may be between $150 and $700/t CO2 removed. These estimates do not account for CO2 emissions from the process or supply chain. Powering an electrochemical system with ~2 MWh/t CO2 removed using current unabated U.S. grid average emissions (400 kg/MWh) would result in 0.8 t CO2 emitted (or a 5× cost increase for net removal).
The technoeconomic assessment of removing CO2 as a gas from seawater through (acid and base) bipolar membrane electrodialysis was undertaken by Eisaman et al. (2018), who estimated costs on the order of $373–$2,355/t CO2 removed. This included an acid stand-alone process ($1,839–2,355/t CO2), an acid process colocated with seawater cooling ($727–$1,076/t CO2), or an acid process colocated with desalination ($436–$717/t CO2). Costs were also included for a base stand-alone process ($1,076–$1,349/t CO2), a base process colocated with seawater cooling ($583–$899/t CO2), or a base process colocated with desalination ($373–$604/t CO2).
The above costs (summarized in Figure 8.13) do not include those associated with monitoring, verification, and reporting. For those processes that remove CO2 from seawater for injection and storage into geological reservoirs, costs are incurred for seismic baseline monitoring and the installation of monitoring wells prior to injection (both of which are initial capital expenditures). Additional geophysical monitoring, water column seawater chemistry analysis, and groundwater monitoring are required during and following (~30 years) injection. However, the estimated costs of this are relatively small <$1/t CO2 (IPCC, 2005). A monitoring program for well-mixed effluent quality will be independent of volume, and daily water sampling and analysis may cost on the order of $50–$100K/yr (<1 percent of the operating costs of the systems in Table 8.4). A more comprehensive environmental monitoring program and environmental impact assessment may form the basis of obtaining a permit for operating a plant within national jurisdictions (e.g., discussed in Chapters 2 and 9).
Governance
The legal framework for ocean CDR is discussed in Chapter 2. Many of the international and domestic laws discussed in that chapter could apply to ocean CDR electrochemical engineering, and specific issues associated with OAE are discussed in Chapter 7.
Previous research (Webb et al., 2021) has considered the application of international and domestic law to electrochemical approaches involving alkalinity creation. Webb et al. (2021) concluded that projects that employ alkalinity creation approaches for the purpose of mitigating climate change would likely constitute “geoengineering” for the purposes of Decisions X/33, XI/20, and XIII/4 under the Convention on Biological Diversity (CBD).10 The decisions recommend that parties to the CBD and other governments avoid geoengineering activities that may affect biodiversity,
___________________
10 Report of the Conference of the Parties to the Convention on Biological Diversity on the Work of its Tenth Meeting, Decision X/33 on Biodiversity and Climate Change, Oct. 29, 2010 (hereinafter Decision X/33)]; Report of the Conference of the Parties to the Convention on Biological Diversity on the Work of its Eleventh Meeting, Decision XI/20 on Climate-related Geoengineering, Dec. 5, 2012 (hereinafter Decision XI/20); Report of the Conference of the Parties to the Convention on Biological Diversity on the Work of its Thirteenth Meeting, Decision XIII/4, Dec. 10, 2016 (hereinafter Decision XIII/4).
TABLE 8.4 A Simplified Techno-Economic Overview of Diaphragm and Membrane Chlor-alkali Systems, and Then the Application of the Produced NaOH for CDR by Ocean Alkalinity Enhancement
| Diaphragm | Membrane | |
|---|---|---|
| System size (kt/yr) | ||
| Chlorine production | 179 | |
| Sodium hydroxide production | 197 | |
| Hydrogen production | 5 | |
| Maximum CO2 removal | 286 | |
| Power (MW) | 71 | 79 |
| Capital costs (M$) | ||
| Cells | 38 | 73 |
| Rectifiers | 8 | 7 |
| Boiler | 9 | 3 |
| Storage | 12 | 12 |
| Sulfate removal | 1 | 2 |
| Cell renewal | 5 | 5 |
| Initial materials | 61 | 49 |
| Total bare module costsb | 134 | 152 |
| Total production costsb | 154 | 175 |
| Total overnight costsb | 172 | 195 |
| Operating Costs (M$/yr) | ||
| Energy costsc | 118 | 83 |
| Raw material costsc | 14 | 15 |
| Fixed running costsc | 10 | 11 |
| Cost summary | ||
| Levelized cost of CO2 removed ($/t CO2)d | 668 | 533 |
| Possible H2 and Cl2 revenue (M$/yr)e | 14 | 16 |
| Levelized cost of CO2 removed ($/t CO2) including revenue | 608 | 466 |
| Levelized cost of NaOH ($/t) as 50% mass solution, including revenue | 442 | 339 |
a Bare module costs were summarized from Abam Engineers (1980) and converted into current prices using the Chemical Engineering Plant Cost Index (https://www.chemengonline.com). Mass flows were taken from Abam Engineers (1980).
b Assuming 10% engineering procurement and contractor costs, 5% project contingency, 12% owner’s costs.
c Cell power of 7,267–8,105 Mcal/ECU, motor power of 703–735 Mcal/ECU, produced hydrogen used in the boiler. In the diaphragm process, 625 Mcal/ECU of additional fuel is required. Raw material costs include salt ($50/t), sulfuric acid ($100/t), soda ash ($150/t), calcium chloride ($150/t). Labor costs include 40 shift and 25 day workers on an annual salary of $35K; 15% of labor as supervision; 35% of labor as administration and overheads; 2.5%, 1%, and 1% of production costs as maintenance, taxes, and insurance, respectively.
d Assuming a capital charge factor of 7.5%, a plant capacity factor of 90%, an escalation rate of 2%, a discount rate of 8%, and a levelization period of 30 years.
e Cl2 at $80/t and H2 at $800/t.
except for “small scale scientific research studies . . . conducted in a controlled setting.”11 The decisions are not legally binding, however. The CBD itself arguably does not prevent countries from undertaking or authorizing electrochemical engineering projects, provided that they comply with all applicable consultation and other requirements imposed by the Convention (Webb et al., 2021).
Webb et al. (2021) further concluded that electrochemical engineering approaches that involve discharging materials into ocean waters could, in some circumstances, be considered “pollution” of the marine environment under the United Nations Convention on the Law of the Sea (UNCLOS) and/or marine “dumping” under the London Convention and Protocol. As discussed in Chapter 2, the parties to the London Protocol have agreed to an amendment, dealing specifically with the discharge of materials into ocean waters for the purposes of “marine geoengineering.”12 The amendment, which has not yet entered into force, establishes a framework under which parties to the London Convention may approve certain marine geoengineering activities involving ocean discharges.13 While electrochemical engineering approaches could be considered a form of “marine geoengineering,” at the time of writing, the framework only applied to activities relating to ocean fertilization. It could be expanded to apply to other techniques in the future (Brent et al., 2019; Webb et al., 2021). However, even if that occurred and the amendment entered into force, it would not be legally binding on the United States, which is not a party to the London Protocol (Webb et al., 2021).
The United States is a party to the London Convention. That instrument requires ocean dumping to be permitted and imposes restrictions on when permits can be issued. Webb et al. (2021)
___________________
11 Para. 8(w), Decision X/33; Para. 1, Decision XI/20; Preamble, Decision XIII/4.
12 Resolution LP.4(8), Amendment to the 1996 Protocol to the Convention on the Prevention of Marine Pollution by Dumping of Wastes and Other Matter, 1972 to Regulate Marine Geoengineering, Oct. 18, 2013.
13 Annex 4, Resolution LP.4(8).
concluded that parties to the London Convention could issue permits for ocean CDR electrochemical engineering approaches involving the production of alkalinity, at least in some circumstances.
Webb et al. (2021) also reviewed the U.S. environmental laws potentially applicable to ocean CDR electrochemical engineering approaches involving the production of alkalinity. They concluded that the applicable laws will depend on the precise nature and location of each electrochemical engineering project. For example, projects involving the installation of structures in U.S. waters may require approvals from the U.S. Department of the Interior Bureau of Ocean Energy Management, U.S. Army Corps of Engineers, and/or other government agencies. Additionally, permits from the U.S. Environmental Protection Agency or state environmental agencies may be required for projects involving the discharge of materials into ocean waters, depending on where they occur. Projects in which CO2 is injected into the sub-seabed could present additional legal challenges (see, e.g., Webb and Gerrard, 2018, 2019).
8.6 SUMMARY OF CARBON DIOXIDE REMOVAL POTENTIAL
The criteria for assessing the potential of electrochemical processes as a feasible approach to ocean CDR, described in Sections 8.2–8.5, is summarized in Table 8.5.
8.6 RESEARCH AGENDA
Components
Ocean-based electrochemical CDR approaches are promising in that (1) technologies that already exist (e.g., for alkaline electrolysis, in the chlor-alkali industry, and to a much smaller extent, involving electodialysis applications) could be adapted for the purpose, (2) existing technologies have previously been scaled to ~0.1 Gt/yr of products over several decades suggests scalability of new technologies, and (3) the environmental impact of the approaches that discharge effluent to ocean can be relatively easily monitored. However, electrochemical approaches present considerable challenges. For most proposed approaches, even best-case cost estimates are greater than other CDR approaches, which are primarily a result of capital expenditure and energy requirements. Given the energy requirements, these processes are unlikely to be feasible for CDR without exploiting low-carbon electricity. Some proposals generate chlorine and/or acids, which require treatment or disposal. An upscaled industry will likely require the movement of large volumes of seawater through the reaction system. Some of these proposals increase ocean alkalinity, and will share a research agenda that considers the wider social, environmental, and governance issues (Chapter 7). Table 8.6 presents the research agenda for advancing understanding of the feasibility of using electrochemical approaches to durably remove atmospheric CO2.
Cost Discovery and Demonstration
Most processes have yet to be explored beyond bench-scale systems and/or early-stage technoeconomic analysis. While useful in scoping the possible range of costs, accurate assessment of first-of-a-kind systems requires the development of pilot and demonstration facilities approaching scales on the order of tonnes per day. For accurate cost discovery, exploration of these at a scale of, at minimum, hundreds of tonnes per year is required.
TABLE 8.5 CDR Potential of Electrochemical Processes
| Knowledge base What is known about the system (low, mostly theoretical, few in situ experiments; medium, lab and some fieldwork, few carbon dioxide removal (CDR) publications; high, multiple in situ studies, growing body of literature) |
Low–Medium Processes are based on well-understood chemistry with a long history of commercial deployment, but is yet to be adapted for CDR by ocean alkalinity enhancement (OAE) beyond benchtop scale. |
| Efficacy What is the confidence level that this approach will remove atmospheric CO2 and lead to net increase in ocean carbon storage (low, medium, high) |
High Confidence Monitoring within an enclosed engineered system, CO2 stored either as increased alkalinity, solid carbonate, or aqueous CO2 species. Additionality possible with the utilization of by-products to reduce carbon intensity. |
| Durability Will it remove CO2 durably away from surface ocean and atmosphere (low, <10 years; medium, >10 years and <100 years; high, >100 years), and what is the confidence (low, medium, high) |
Medium–High >100 years Dynamics similar to OAE. |
| Scalability What is the potential scalability at some future date with global-scale implementation (low, <0.1 Gt CO2/yr; medium, >0.1 Gt CO2/yr and <1.0 Gt CO2/yr; high, >1.0 Gt CO2/yr), and what is the confidence level (low, medium, high) |
Medium–High Potential C removal >0.1–1.0 Gt CO2/yr (medium confidence) Energy and water requirements may limit scale. For climate relevancy, the scale will need to be doubled to an order of magnitude greater than the current chlor-alkali industry. |
| Environmental risk Intended and unintended undesirable consequences at scale (unknown, low, medium, high), and what is the confidence level (low, medium, high) |
Medium–High (low confidence) Impact on the ocean is possibly constrained to the point of effluent discharge. Poorly-known possible ecosystem impacts similar to alkalinity enhancement. Excess acid (or gases, particularly chlorine) will need to be treated and safely disposed. Provision of sufficient electrical power will likely have remote impacts. |
| Social considerations Encompass use conflicts, governance-readiness, opportunities for livelihoods, etc. |
Similar to OAE and to any industrial site. Substantial electrical power demand may generate social impacts. |
| Co-benefits How significant are the co-benefits as compared to the main goal of CDR and how confident is that assessment |
Medium–High (medium confidence) Mitigation of ocean acidification; production of H2, Cl2, silica. |
| Cost of scale-up Estimated costs in dollars per metric ton CO2 for future deployment at scale; does not include all of monitoring and verification costs needed for smaller deployments during R&D phases (low, <$50/t CO2; medium, ~$100/t CO2; high, >>$150/t CO2) and confidence in estimate (low, medium, high) |
High >$150/t CO2 (medium confidence) Gross current estimates $150–$2,500/t CO2 removed. With further R&D, it may be possible to reduce this to <$100/t CO2. |
| Cost and challenges of carbon accounting Relative cost and scientific challenge associated with transparent and quantifiable carbon tracking (low, medium, high) |
Low–Medium |
| Cost of environmental monitoring Need to track impacts beyond carbon cycle on marine ecosystems (low, medium, high) |
Medium (medium–high confidence) All CDR will require monitoring for intended and unintended consequences both locally and downstream of CDR site, and these monitoring costs may be substantial fraction of overall costs during R&D and demonstration-scale field projects. |
| Additional resources needed Relative low, medium, high to primary costs of scale-up |
Medium–High High energy requirements (1–2.5 MWh/t CO2 removed) and build-out of industrial CDR. |
Electrode and Membrane Materials
Novel electrode materials may be able to substantially reduce the generation of Cl2 gas from the anode favoring the production of O2. A research agenda may be able to significantly reduce the cost and improve the technical feasibility of electrochemical approaches by exploring durable, long-lasting, and cost-effective electrode materials and their optimum design and configuration with either membrane-based or membraneless reaction systems.
Acid Management and Environmental Impact Assessment
At scale, the generation of large volumes of Cl2 gas or acids may be unavoidable. If electrochemical approaches are to be deployed at scale, then an important component of a research strategy includes how these wastes can be safely disposed of. Waste management strategies are most effective when they are part of a comprehensive environmental impact assessments or life-cycle assessments that consider not only the impact of the process, but also its supply chain. This work could assess what impacts are possible to mitigate, what operating protocols are needed, and what impacts are likely even in highly controlled systems. It would also be useful to explore worst-case impact scenarios.
Coupling to Whole-Rock Dissolution Systems
Optimum system design will likely require careful coupling between the electrochemical component and that which facilitates rock dissolution. Furthermore, large-scale electrochemical approaches would use a range of local rock resources, and research to assess the impact of real-world rock chemistries on system performance and/or effluent quality is needed, including the potential of using waste rock (e.g., mine tailings, quarry fines, etc.) or alkaline materials (e.g., slag from the steel industry, concrete and demolition waste, etc.).
Hybrid Approaches
Processes that combine CDR and alkalinity increase are sparse in the literature but could be particularly useful in maximizing removal for small changes in system operation.
Resource Mapping and Pathways to Upscaling
It is not yet clear whether or how rapid a large-scale deployment of electrochemical approaches could be achieved. Research is needed to explore pathways and incentivization
strategies for scale-up. These pathways could include integration within existing industries (e.g., desalination, power station cooling water, etc.), which may provide the most feasible, albeit niche, deployment for first-of-a-kind systems. It could also be possible to integrate products from these electrochemical systems into existing markets (e.g., hydrogen, chlorine, oxygen, silica) to lower the cost of initial systems. It is possible that the interaction between a growing electrochemical industry (and its supply chain) and existing industry, economies, and markets may have unintended consequences. A resource mapping exercise that identifies the colocation of low-carbon electricity, concentrated brine feedstocks, rock extraction, seawater, CO2 disposal, and product utilization would be essential in designing upscaling pathways. This activity should be undertaken with stakeholders in the supply chain.
Cost and Time Frame of Research Agenda
Demonstration projects could be commissioned relatively quickly (~24 months) and would require a similar operational time in which to test the systems. These would be most effective if they were underpinned by smaller-scale laboratory-based experimentation and materials and systems development. Multiple rounds of demonstrations over a 5- to 10-year time frame would allow for iterative development between stages. Within the initial 36 months, an assessment of resources and upscaling pathways will be critical in assessing the viability of the approaches. Research exploring the environmental impact, and acid management or reduction strategies would support the demonstration research (in the first 60 months), but may not be necessary thereafter. In total the research agenda would cost ~$475 million over a 10-year period
Environmental Impacts of Research Agenda
The research described above could be undertaken with minimal environmental impact, and almost no impact on the ocean. Seawater may need to be collected as a feedstock for the processes (although artificial seawater could also be appropriate in some experiments). Depending on local regulations and the scale of the experiment, process effluents (i.e., brine, acids, and bases) could be neutralized and or diluted prior to disposal in municipal sewer systems. Volumes of solid wastes would be small and relatively inert. The research agenda described above considers the systems required to remove CO2 from seawater, or to increase its alkalinity. The wider ocean impacts from ocean alkalinity enhancement are included in Chapter 7.
8.7 SUMMARY
The research agenda for electrochemical processes focuses on activities that reduce the cost and environmental impact of the approaches. Presently, only a small number of system configurations have been investigated for either CDR from seawater or as methods for increasing ocean alkalinity. These should be broadened to also consider novel designs of electrolyzers and electrochemical reactors, electrode materials that can minimize the production of unwanted by-products (e.g., Cl2 gas), electrochemical reactor architectures, and hybrid approaches that both remove CO2 and increase ocean alkalinity. Wider impacts on ocean ecosystems are included in the research agenda for Ocean Alkalinity Enhancement and are not repeated here. We recommend that meaningfully large demonstration-scale projects (>1,000 kg/day, and approaching the scale of several to tens of tonnes per day) be central to the research agenda. While part of this research agenda may focus on the development of specific technologies or materials (e.g., electrolyzer design or novel electrode
TABLE 8.6 Research and Development Needs: Electrochemical Processes for Ocean Alkalinity Enhancement
| # | Recommended Research | Gap Filled | Environmental Impact of Research | Estimated Research Budget ($M/yr) | Time Frame (years) |
|---|---|---|---|---|---|
| 8.1 | Demonstration projects including CDR verification and environmental monitoring | What is the likely upscaled cost of the approach? | Low | 30 | 5 |
| 8.2 | Development and assessment of novel and improved electrode and membrane materials | What is the minimal production possible of Cl2 (or other waste gases) from electrochemical systems? Can membranes be optimized for the unique constraints encountered in electrochemical ocean CDR approaches? | Low | 10 | 5 |
| 8.3 | Assessment of environmental impact and acid management strategies | What is the life-cycle environmental impact of approaches? | Low | 7.5 | 10 |
| 8.4 | Coupling whole rock dissolution to electrochemical reactors and systems | Is it possible to scale electrochemical approaches while managing waste acid production? | Low | 7.5 | 10 |
| 8.5 | Development of hybrid approaches | Can approaches be optimized or combined for cost reduction? | Low | 7.5 | 10 |
| 8.6 | Resource mapping and pathway assessment | Do we have sufficient resources in the right locations to upscale production? | None | 10 | 5 |
NOTE: Bold type identifies priorities for taking the next step to advance understanding of electrochemical processes as an ocean CDR approach.
materials), it should also focus on systems integration (particularly with rock dissolution processes) and scale-up strategies that allow for meaningful process cost reductions to be discovered.
The processes explored by the research agenda will need to demonstrate pathways to large-scale deployment. As such, the research agenda should also include provision for resource mapping, stakeholder engagement, and wider economic impact from an ocean-based electrochemical CDR industry operating at a climate-relevant scale. The outcomes of the research should be transparently disseminated such that independent assessment of cost and environmental impact can be made. Social and governance considerations should also be addressed in the research.






























