
Appendix H
‘Omics Techniques
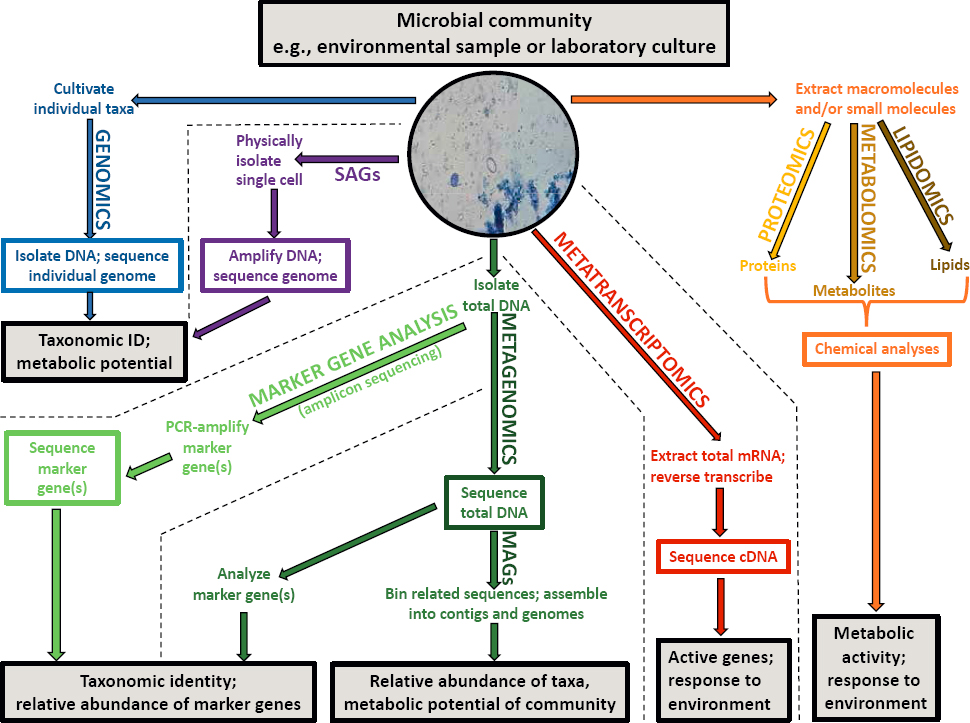
The following text provides an overview of ‘omics techniques, focusing on their application to analysis of microbial communities present in environmental samples or laboratory cultures. Note that ‘omics techniques may be used differently to study individual higher organisms and their structures or organs (see Chapter 6).
Genomics is the sequencing and study of genomes (i.e., the full complement of DNA in cells, including chromosomes, plasmids, and virus genomes) including genome structure (e.g., chromosome sequence and gene order), function (e.g., potential biochemical activity) and evolution (e.g., mutability and genetic exchange). Genomes are
SOURCE: Inset micrograph adapted from Reef2Reef.
obtained by sequencing the entire DNA content of a single organism or colony of identical cells then, ideally, organizing those sequences into a complete genome without sequence gaps. The genome sequence can reveal the taxonomy (classification) of an individual organism, potential enzymatic capabilities (based on gene sequences that encode enzymes or structural proteins), as well as mobile accessory genes encoded on plasmids or viral genomes (e.g., bacteriophage). The sequences encoding functional or structural products (e.g., proteins, RNA molecules used in protein synthesis and DNA replication) are commonly identified by comparison to publicly available databases using bioinformatics tools. For example, the genomes of hydrocarbon-degrading bacteria contain suites of genes encoding enzymes (catalytic proteins) that act sequentially in biochemical pathways that degrade specific hydrocarbon classes. The genomes usually contain all the genes encoding enzymes for a complete biodegradation pathway plus the regulatory genes that control when those enzymes are produced (see Transcriptomics below) as well as structural proteins for solute transport, detoxification, and sometimes motility (see Proteomics and Metabolomics below). Comparative genomics enables the discovery of novel genes in uncultivated organisms by comparison with known organisms. Pairing genomics information with classical cultivation methods and metabolomics strengthens the use of certain genomes (or selected marker genes) as proxies for biodegradation potential in a given environment.
An exciting development in genomics is the single-cell amplified genome (SAG) technique that, as the name implies, allows the genome of an isolated individual microbial cell to be sequenced without requiring cultivation of the cell. A microfluidic device is used to physically manipulate individual cells (e.g., suspended in seawater) into microscopic wells where the cell is lysed and its DNA amplified, sequenced, and assembled into contigs (contiguous DNA sequences assembled bioinformatically by overlapping multiple shorter DNA sequences) and scaffolds (long sequences comprising contigs and gaps). If sufficient sequencing “coverage” of the genome has been achieved, it is possible to assemble the contigs and scaffolds into a complete genome, but often there are gaps and the virtual organism is delineated by the inferred percentage of completeness of the genome. The process is facilitated if there is an identical or very similar genome already in a database for comparison, but it is also possible to generate a virtual genome de novo (i.e., solely from overlapping sequenced fragments of a hitherto-unknown organism). In this way, key microbes that cannot yet be cultivated in a laboratory for characterization can be sequenced and their activity inferred from their genome sequence. For obligate symbionts that cannot be cultivated alone and/or may be present in small proportions in a community, such as those implicated in anaerobic hydrocarbon biodegradation, SAG is a breakthrough technique.
Metagenomics is the study of the collective genomes comprising a mixed community of organisms, such as the microbiota in an environmental sample or a laboratory culture containing multiple species. First, the cells are lysed to release total community DNA (a step that introduces potential bias). This DNA can then be used in several ways to acquire different information: (1) Marker gene sequencing and analysis of bacterial 16S rRNA gene regions is commonly used to determine the taxonomic types and relative proportions of different microbes within a community. A specific region of this marker gene is amplified from the extracted DNA using polymerase chain reaction (PCR) then sequenced by high-throughput sequencing (aka next-generation sequencing). Various bioinformatic tools remove spurious sequences, ensure quality control, and compare the marker gene sequences to public databases such as GenBank, SILVA, RDP, or Greengenes to infer the identity of discrete groups of sequences (taxa) based on their similarity to archived sequences and their place in phylogenetic trees constructed using the same marker gene. The reference sequences may be from cultivated isolates that have been studied and characterized, or may themselves be known only as a DNA sequence (an operational taxonomic unit or amplicon sequence variant) that was also obtained by sequencing community DNA and has never been cultivated in a laboratory. In the latter case, the metabolism and behavior of the “virtual microbe” can only be inferred through its relationship to the relatively few cultivated organisms that have been studied biochemically in a lab. Although conventional (non-quantitative) rRNA gene analysis does not yield information about the absolute cell numbers of different taxa in the sample, analysis of multiple samples can reveal the taxonomic diversity of the community as well as changes in community composition that occur over time and space or in response to changing environmental conditions, such as pre- and post-exposure to oil. Quantitative PCR allows estimation of numbers of marker genes in a sample Notably, PCR amplification is known to introduce biases that can over- or under-represent particular taxa. This is one of the reasons why marker gene sequencing, which has been used extensively in the past two decades, is largely being displaced by (2) shotgun metagenome sequencing. In this technique, the entire community DNA is sequenced without PCR amplification and often without sequence assembly, and desired marker genes are sorted in silico by bioinformatic software. Selecting and analyzing taxonomic marker sequences like 16S rRNA genes will reveal the diversity of the community and the relative abundance of taxa, whereas analysis of functional marker genes (e.g., those encoding key enzymes) by using annotation software and functional gene databases can reveal the presence, diversity, and relative abundance of genes associated with potential metabolic activity if those genes are expressed (e.g., hydrocarbon biodegradation). Alternatively, using bioinformatics software the sequence information can be sorted into “bins” of related sequences that are then reconstructed into (3) metagenome-assembled genomes representing individual taxa in the community.
This generates individual genomes (complete or partial) for further study and inference. One benefit to adding assembly to the analysis is that there is context for the marker genes (e.g., associating a potential enzyme activity with a specific biochemical pathway or a particular taxon). Metagenomic analysis of samples collected at different stages of oil biodegradation can reveal which species are affiliated with biodegradation of certain oil components as well as the potential trajectory of that biodegradation activity (e.g., partial or complete oxidation of the hydrocarbons).
It is important to note that the presence of a DNA sequence in a (meta)genome, for example one encoding an enzyme, is only an indicator of the potential activity of the organism; it does not necessarily mean that the organism (or community) is actually producing that enzyme or exhibiting the biochemical function encoded by that sequence. To determine whether the genetic potential is being manifested is informed by transcriptomics, the study of the mRNA transcripts in an organism (i.e., the expression of the complement of its genes that are active under specific conditions). This is achieved by sequencing the total mRNA (or, more precisely, its complementary DNA) present in the cell at a specific sampling time. A microbe may only express (transcribe) genes for a particular biochemical pathway if the substrate for that pathway is present in the environment; in its absence, the genes are still present in the genome and would be detected in the genomic sequence as potential activity, but they are silent and will not contribute to the transcriptome. The community extension of transcriptomics is the field of metatranscriptomics, the study of the full complement of DNA transcripts in a community of organisms. This method was developed to study the aggregate gene expression of all members of the community under specific conditions. This is important because the transcriptome of a single species in isolation (e.g., in pure culture in a laboratory) likely will differ from its expression in the environment and in the presence of a community, because biochemical signals from the whole can enhance or suppress gene expression in individual community members. Metatranscriptomic studies often examine changes in transcription under two or more different conditions rather than just in a given baseline condition. That is, it is the relative abundance of transcripts, both up- and down-shifted, that provides information about how the community responds to environmental conditions such as oil incursion or oxygen depletion. Combining metatranscriptomic and metagenomic analyses yields a fuller picture of how a community behaves under baseline and perturbed conditions, something that was impossible only a decade ago.
Proteomics, lipidomics, and metabolomics study the products of gene transcription—that is, which macromolecules (proteins and lipids) and metabolites the organism(s) produce under specific conditions. Typically, these studies are conducted under two or more different growth conditions to highlight changes in cell or community response to particular stimuli (e.g., temperature, growth substrates, toxins, etc.) and, as with transcriptomics, the data are often reported as fold increases or decreases from a baseline condition. Advanced analytical chemistry techniques such as mass spectrometry and nuclear magnetic resonance are used to characterize the proteome, lipidome, and/or metabolome of organisms by comparison to public databases (e.g., UniProt, LipidMaps, and National Metabolomics Data Repository, respectively) and to infer biochemical pathways and metabolic products.
Proteomics is the study of the total complement of proteins (the proteome) produced by an organism under specific conditions. It includes protein identification, quantification, and enzymatic modification of proteins and is commonly studied using mass spectrometry of protein extracts. The proteins may be catalytic (e.g., enzymes), structural (e.g., proteins for uptake of substrates or excretion of metabolites, or for motility toward substrates like oil or away from toxins), or regulatory (controlling transcription of genes).
Lipidomics is the study of the identity, structure, and function of the complete profile of lipids produced by an organism (the lipidome), such as those comprising cell membranes (and organelles in eukaryotes) that affect surface area for substrate uptake and waste excretion, or for specialized functions like photosynthesis.
Metabolomics is the study of small molecules produced by organisms as intermediates or end products of biochemical pathways (i.e., metabolites). For example, some microbes completely oxidize hydrocarbons to CO2 and H2O, whereas other hydrocarbons are only partially oxidized and the pathway metabolites (e.g., acids or alcohols) are excreted to the external environment.
Metabolism, which typically can only be inferred from taxonomy, is best deduced by comparison to cultivated organisms (a highly biased set, as mentioned above) or by analyzing metatranscriptomes and/or macromolecules. In conjunction with ‘omics, incubating a community with a growth substrate enriched in stable carbon or nitrogen isotopes (13C,15N; stable isotope probing) specifically labels the macromolecules and metabolites of active organism(s) directly involved in substrate assimilation so that key microbial species can be differentiated from “accessory” members of a community. Thus, ‘omics can reveal significant shifts in community composition in response to environmental conditions, can predict and identify metabolic potential, and may provide clues that eventually allow cultivation of “virtual microbes” for detailed biochemical and environmental study.




