
2
Overview of Hereditary Disorders of Connective Tissue
Heritable disorders of connective tissue (HDCTs) are a heterogeneous group of many inherited disorders and subtypes. Two of the most common HDCTs, Marfan syndrome (MFS) and the Ehlers-Danlos syndromes (EDS), are discussed in depth in Chapters 3 and 4, respectively, along with several related disorders. This chapter addresses the history, etiology, diagnosis, and management considerations common to all HDCTs.
HISTORY OF HEREDITARY DISORDERS OF CONNECTIVE TISSUE
Connective tissue—tissue that helps support, protect, and provide structure to other tissues and organs in the body—is the most abundant and diverse of tissues. Connective tissues vary with respect to their type of cells and cellularity (dense versus loose). Specialized connective tissues include bone, cartilage, ligaments, tendons, and adipose tissue (fat). The role of connective tissues in the vascular system, the gastrointestinal system, the intervertebral disc, and muscle is also recognized, and has furthered understanding of HDCT phenotypes and symptomatology.
The term “heritable disorder of connective tissue” was coined by Dr. Victor McKusick, who first wrote about the concept in his 1955 article “The Cardiovascular Aspects of Marfan’s Syndrome: A Heritable Disorder of Connective Tissue” (McKusick, 1955). McKusick’s thinking about HDCTs was influenced by the work of Follis, who studied a case of lethal osteogenesis imperfecta (OI) and demonstrated that the organic matrix of bone and the connective tissue of the skin and sclera were defective (Follis,
1952, 1953a,b). The first edition of McKusick’s Heritable Disorders of Connective Tissue, published in 1956, included discussion of MFS, EDS, OI, pseudoxanthoma elasticum, and Morquio syndrome. By the time the fourth edition was published in 1972, chapters had been added on homocystinuria, alkaptonuria, Menkes disease, and cutis laxa, as well as the skeletal dysplasias or osteochondrodystrophies.
Since McKusick’s initial writings in the 1950s, the concept of HDCTs has become firmly ingrained in the genetic lexicon. The 2002 volume Connective Tissue and Its Heritable Disorders, edited by Royce and Steinmann, includes 26 chapters, many of which subsume multiple distinct diagnoses (for example, Chapter 23 includes the skeletal dysplasias, of which there are now more than 450 well-delineated forms) (Mortier et al., 2019).
Two technological advances—the availability of genomic sequencing and ready access to information through the internet—have forever changed the way the medical community views HDCTs, as well as many other rare disorders. The ability of people affected by rare disorders to comb through online medical information and “ask Dr. Google” for information about their conditions has led to self-diagnosis in many cases. Affected people also are now able to share their experiences with a worldwide population of similarly affected individuals. Patient support groups, such as The Marfan Foundation and The Ehlers-Danlos Society, have encouraged and supported research, including basic, translational, and clinical research, that otherwise might not have been accomplished. Moreover, an increasing willingness, and even urgency, to understand the lived experience of people with these conditions has led to the development of a wide range of patient-reported outcome measures to help inform research on these conditions and the disability they cause.
EPIDEMIOLOGY
HDCTs are a complex group of disorders that can be difficult to diagnose. Since their genetic basis is heterogenous, the incidence of an individual HDCT can range from commonly occurring—for example, hypermobility spectrum disorders (HSD)—to rare recessively inherited disorders. Determining the true incidence of these conditions is challenging because it relies on recognition of the disorders, which is historically biased toward their more severe, and therefore recognizable, expressions, and on retrospective reviews and diagnosis codes.
Since many of these disorders are underappreciated, their precise prevalence is not established. For example, MFS is estimated to occur in 1 to 5 per 10,000 individuals (Judge and Dietz, 2005; NORD, 2021), while the prevalence of the phenotypically overlapping disorder Loeys-Dietz syndrome is
unknown (Loeys and Dietz, 2018). The prevalence of HSD and different EDS types also varies significantly. Current research indicates that the EDS are thought to occur in about 1 in 5,000 people (Pyeritz, 2000; Steinmann et al., 2002). Among the EDS, hypermobile EDS (hEDS) is the most common type, while other types are much rarer. The estimated prevalence of vascular EDS, for example, is between 1/50,000 and 1/200,000 (Byers, 2019), while that of both musculocontractural EDS and dermatosparaxis EDS is less than 1/1,000,000 (Orphanet, 2022a,b).
GENETICS
For many years, the term “mutation” was used to describe changes in genes resulting in heritable conditions. As a result of the Human Genome Project and ready access to rapid genomic sequencing, new language has been adopted to describe the changes observed in human DNA; such changes are now referred to as “variants.” The classification of sequence variations has been standardized according to guidelines published by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (Richards et al., 2015). It is recognized that some differences among the DNA sequences of humans have no discernable phenotypic consequences; such changes are termed “benign.” Other changes are unequivocally disease-causing and are referred to as “pathogenic.” Between these extremes are variants that are categorized as “likely benign” or “likely pathogenic.” In addition, a wide array of variants are called “variants of unknown significance” because current knowledge is insufficient to classify them as benign or pathogenic. Study of the different types of variants in the human DNA sequence is ongoing, and variants are frequently reclassified as more information becomes available.
The concept of pleiotropism is central to the study of genetic disorders and is a universal feature of HDCTs. The term is used to describe how variation in a single gene may manifest in multiple organ systems. The specific organs affected by such a variation will be determined by the pattern of the gene’s expression during embryological development, postnatal growth, and maintenance of tissue. For example, type I collagen is expressed in bone, skin, tendons, and ligaments; all of these tissues are impacted by the pathogenic variants of type I collagen that cause some forms of OI.
Variable expressivity, another standard concept in medical genetics, is also a hallmark of the HDCTs. This term denotes the different degrees of severity that may be seen in individuals carrying the same pathogenic DNA variant, even among members of the same family. Much work is still necessary to develop a full understanding of the gene–gene and gene–environment (e.g., tobacco use) interactions that contribute to the wide variation in expression seen in Mendelian genetic disorders.
CONNECTIVE TISSUE
Connective tissue is an integral component of every organ system and plays a crucial role in their function. Hence the physical and mental secondary impairments associated with HDCTs, which may develop and progress over time, manifest throughout the body and affect functioning in every body system. As disorders of connective tissue, HDCTs share many features because the molecules or genetic variants that produce disease are expressed in overlapping tissues. Thus, joint issues are seen in MFS, EDS, congenital contractural arachnodactyly (CCA), and the skeletal dysplasias since the respective genes that lead to these diseases are expressed in the tissues of the joints, as well as in other tissues. HDCTs arise from defects in the genes that encode extracellular matrix (ECM) molecules or molecules related to matrix biosynthesis and cell signaling. These molecules provide the physical structure for complex connective tissues, and are critical for appropriate physiological functions as they also dictate the functions of the cells within the tissues. There are two types of ECM—interstitial and pericellular. The interstitial matrix interconnects cells in connective tissues, while the pericellular matrix is a unique cell-adjacent matrix. The composition of the ECM varies among tissues based on each tissue’s specialized functions. Bone ECM, for example, strongly expresses type I collagen, whereas in cartilage ECM, type II collagen predominates, and type I collagen is present at relatively low levels.
The components of connective tissue include cells, fibers, and ground substance (Nezwek and Varacallo, 2021). Figure 2-1 depicts the structure of connective tissue, which includes cells with receptors, such as integrins; fibers, such as collagen and elastin; and ground substances, such as proteoglycans. Ground substance is the amorphous gelatinous material in which the cells and fibers are embedded. It is composed primarily of water, along with hyaluronic acid, proteoglycans, and proteins (e.g., laminin and fibronectin) that function as glue for the cells in the ECM (Nezwek and Varacallo, 2021). This ground substance allows for the exchange of nutrients between cells and capillaries. Three basic types of fiber make up connective tissue: collagenous fibers, predominantly type I collagen that provides tensile strength to loose and dense connective tissues (Ricard-Blum, 2011); thin reticular fibers, composed of type III collagen that forms cross-links to generate a supportive mesh for tissues (Hayakawa et al., 1990); and branching elastic fibers, composed of elastin that allows tissues to stretch and recoil (Uitto, 1979).
Connective tissues help define the function of specialized tissues. Bone is composed of specialized cells that include osteoprogenitor cells, osteoblasts, osteocytes, and bone-lining cells. Ninety percent of bone ECM is composed primarily of type I collagen; the other 10 percent consists of
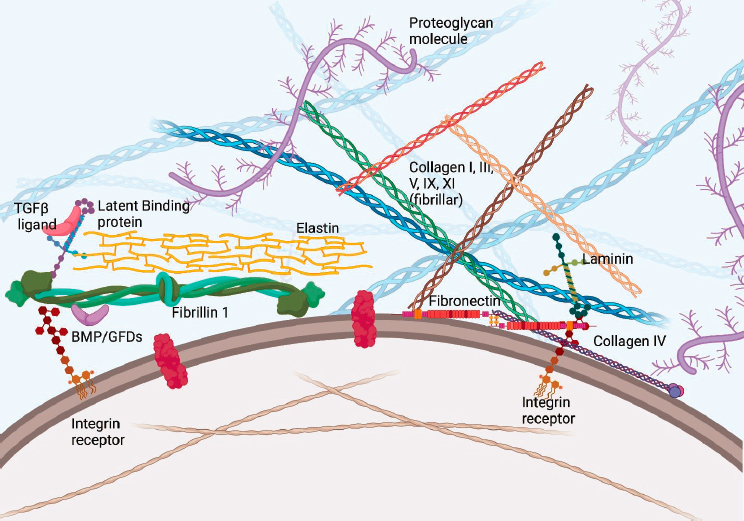
SOURCE: Generated by the committee.
noncollagenous proteins (Paiva and Granjeiro, 2017). In contrast with other connective tissues, bone ECM is mineralized, giving it strength and rigidity to sustain mechanical forces (Weatherholt et al., 2012).
Cartilage has multiple roles, including providing a template for linear growth and bone development, as well as covering for the surface of joints so bones can slide over each other. It comprises cartilage cells (chondrocytes) and a glycoprotein matrix supported by collagen fibers, predominantly type II collagen. Like bone, cartilage is a tough tissue that can withstand forces.
Tendons and ligaments are fibrous and dense connective tissues that connect muscle to bone (tendon) and bone to bone (ligament); they serve to stabilize the skeleton and allow movement by transmitting the mechanical force of muscle contractions to the bone (Ashara et al., 2017; Connizzo et al., 2013). Tendons and ligaments contain fibroblast-like cells, termed tenocytes, or ligament fibroblasts (Kannus, 2000). These cells are located between parallel chains of collagen fibrils. While these collagens are not specific to tendon and ligament, the protein tenomodulin is specifically expressed in these tissues (Shukunami et al., 2006).
Other musculoskeletal connective tissues, including muscle and fascia, have distinct ECMs that contribute to their function. Muscle fibers, made up of myocytes and progenitor cells, are embedded in a complex meshwork
consisting of collagens, predominantly types I and III; glycoproteins; proteoglycans; and elastin. Muscle ECM plays an important role in development, as well as in muscle fiber force transmission, maintenance, and repair (Gillies and Lieber, 2011). Fascia is a generic term for a continuum of multiple types of connective tissue structures that traverse the entire body, from the surface to deeper (interior) anatomical layers. In recent years, it has been recognized that fascia is more than a packaging unit for the skin, muscle, and other organs. Like other connective tissue structures, it serves as a medium for transmitting forces, particularly those that come from muscle, and it is composed of cells, ground substance, and fibers that form a three-dimensional network. Fibroblasts and fibrocytes are the major cell types in fascia, which is composed predominantly of type I and III collagens, and proteoglycans and glycoproteins contribute to the formation of the ground substance. Newly identified cells in the fascia—telocytes and fasciacytes—are deemed responsible for the fascia’s ability to glide (Dawidowicz et al., 2015; Stecco et al., 2018).
While the vascular system is not considered a connective tissue, the ECM that surrounds and is incorporated into the blood vessels plays an important role in multiple processes associated with their structure and function (del Monte-Nieto et al., 2020). The cardiovascular ECM is formed from more than 300 proteins that include collagens, elastins, fibulins, and laminins, among others. Importantly, the essential mechanical or viscoelastic properties of the vessels are provided by three main constituents: elastic fibers, fibrillar collagens, and large aggregating proteoglycans (Barallobre-Barreiro et al., 2020). The cardiovascular ECM is a highly dynamic structure, and alterations to these ECM molecules can affect the integrity of the cardiovascular system.
Connective tissue is also a structural and functional component of the central and peripheral nervous systems. The meninges provide a fibrovascular and ligamentous suspension system for the brain, the spinal cord, and vascular structures within the cranium and the spine, anchoring the cranial and spinal nerves as they merge with the peripheral nervous system. The meninges also have a role in the production and circulation of cerebrospinal fluid (CSF). The main functions of CSF are to provide a mechanical barrier against shock to the brain and spinal cord, remove metabolic waste products, and transport signaling neurotransmitters. The peripheral nerve fibers, made up of axons, are embedded in a complex framework of fibrocollagenous sheaths, also known as epi-, peri-, and endoneurium. These fibers form a network of corpuscle-like myelinated and unmyelinated axons and structured nerve endings that terminate in the connective tissue layers of the skin, the musculoskeletal system, and the ligaments and joints, where they are variably organized by collagen fibers and contiguous with collagen. The ultrastructure and function of those Ruffini bodies, Pacinian corpuscles,
and Golgi or Merkel receptors have been widely studied, and shown to provide the nociceptive, mechanoreceptive, and proprioceptive network of the human body (Halata, 1977; Vandenabeele et al., 1997; Watanabe et al., 2004). Those units also harbor unmyelinated free-ending axon bundles having direct contact and an intricate relationship with individual collagen fibrils (Abdo et al., 2019).
The gastrointestinal tract consists of four layers—mucosa (innermost layer), submucosa, muscularis propria, and adventitia or serosa—the structure of which varies with their function in different areas throughout the system (University of Leeds, 2022). The submucosa and adventitia layers consist of loose connective tissue that contains blood vessels, lymphatic vessels, and nerves. The submucosa may also contain mucus-secreting cells. The mucosa itself comprises three layers: the epithelial lining; the lamina propria, a layer of loose connective tissue containing vasculature for the epithelium and often mucosal glands, lymphoid follicles, and plasma cells; and the muscularis mucosa, a double layer of smooth muscle (University of Leeds, 2022). Highly vascularized, the gastrointestinal tract is an interface of the nervous and immune systems and represents a confluence of processes impacted by many connective tissue disorders.
Cells of the immune system, such as lymphocytes, mast cells, and macrophages, are found within connective tissue. As the first line of defense in fighting illness and disease, the cells residing in the connective tissue in an activated or inactivated state account for a major part of the entire immune system (Krakower, 1972).
As noted above, genes encode the proteins that constitute the elements of connective tissue. While all connective tissues have the general types of ECM proteins (e.g., collagens, elastin, fibrillin) in common, their modifying genes, ECM organization, and differential gene expression contribute to variation in their function and in their presentation and phenotype in patients. In general, proteins that are more highly expressed in connective tissue have greater influence on tissue properties. Levels of gene expression can influence early development (embryogenesis), ongoing tissue maintenance, and the aging process as gene expression changes (Glass et al., 2013; Işıldak et al., 2020). Furthermore, gene variants can produce different effects in individuals and their connective tissues. For example, variants in a highly expressed protein in a connective tissue may produce mild to severe complications depending on the individual, and the same variant can produce different physical complications among family members. The interactions among genes, the proteins they encode, and environmental factors also can influence the behavior of connective tissues. For example, smoking negatively impacts the expression of type I and III collagen in skin and alters the balance of ECM turnover (Knuutinen et al., 2002), and may further contribute to disease progression in an at-risk individual with an HDCT.
Pathogenic variants in the genes encoding the structural constituents of connective tissue may result in HDCTs, with demonstrable abnormalities in the affected tissues. MFS, for example, results from pathogenic variants in the gene encoding fibrillin-1; this protein is expressed in microfibrils providing structural support in the lung, blood vessels, skin, ciliary zonules, tendon, cornea, and glomerulus. These microfibrils provide a framework for elastin deposition at the periphery of elastic fibers (Jensen and Handford, 2016). Thus, individuals with MFS manifest symptomatology and complications in fibrillin-1–expressing tissues. Examples include increased incidence of spontaneous pneumothoraxes, aortic root dilation with aneurysms, ectopia lentis, joint hypermobility, and renal cysts (Arnaud et al., 2021; Milewicz et al., 2021). Pathogenic variants in the fibrillin-1 gene can produce multiple other disorders as well, including those associated with short stature and limited joint mobility, such as Weill-Marchesani syndrome; geleophysic dysplasia; acromicric dysplasia (Sakai and Keene, 2019); and stiff skin syndrome—a rare disorder that presents in infancy or early childhood and is characterized by rock-hard skin, limited joint mobility, and mild hypertrichosis in the absence of other visceral involvement (Liu et al., 2008; Loeys et al., 2010).
Cartilage has a distinct cell type, the chondrocyte, that contributes to linear growth through a process of recruitment, proliferation, hypertrophy, and transition to bone formation. The chondrocyte also facilitates mobility by creating a low-friction environment and cushioning movement (Sophia Fox et al., 2009). The predominant collagen in cartilage is type II (Omelyanenko and Slutsky, 2014), which is also highly expressed in the vitreous of the eye (Deemter et al., 2009). Type II collagen is encoded by the COL2A1 gene. HDCTs that include a spectrum of skeletal disorders due to pathogenic variants in COL2A1 have significant cartilage involvement. Affected individuals often have significantly shorter stature, early-onset osteoarthritis, and retinal complications due to an abnormal vitreous (Savarirayan et al., 2019). These examples highlight that expression of an abnormal or decreased amount of protein in connective tissues that rely on a functional protein produces phenotypes that affect individuals in multiple organ systems.
DIAGNOSIS
As with many diseases, establishing an accurate diagnosis is an important first step in comprehensive care for individuals living with an HDCT. A specific diagnosis is key to understanding the expected natural history of the individual’s condition, and allows for rational and informed decision making by the patient and clinician, anticipatory management, and optimized outcomes. In general, HDCTs are diagnosed through a combination
of clinical findings, established clinical criteria, and family history, followed by confirmatory molecular genetic testing when the genes responsible for the suspected disorder have been identified.
For those HDCTs for which responsible genes and pathogenic variants have been identified, genetic testing is very specific and can establish a diagnosis. In some cases, genetic testing identifies “variants of uncertain significance” (VUS) that are not diagnostic in and of themselves. They have to be considered in a broader context, and no clinical decisions or genetic counseling should be based solely on the finding of a VUS. For some of the HDCTs for which a molecular cause is known, there is a small percentage of patients and families in whom pathogenic variants are not found. An example is Stickler syndrome, for which at least four different genes have been identified, but a small percentage of families with clinically diagnosed Stickler syndrome will not have identifiable variants in any of the known genes. Notably, the most common HDCTs—HSD/hEDS—currently have no known causative genes, so no genetic testing is available for these disorders.
A combination of clinical findings should lead the informed clinician to suspect a specific HDCT; for example, tall stature and arachnodactyly (long fingers and toes) should lead to a suspicion of MFS or Loeys-Dietz syndrome (LDS); generalized joint hypermobility in conjunction with significant skin hyperextensibility or fragility should lead to a suspicion of one of the varieties of EDS; and a personal or strong family history of early osteoarthritis is suggestive of Stickler syndrome, especially if the family history includes cleft palate, retinal detachment, and/or hearing loss. The first step in establishing a diagnosis is for the clinician to consider the possibility of an HDCT. Once that suspicion has been entertained, clinical algorithms and diagnostic criteria can help provide a specific diagnosis.
For most of the HDCTs, once a clinical diagnosis has been suspected or established, it can be confirmed through molecular genetic testing. At present, genetic testing is performed using a panel of genes that covers a wide range of potential causes for the phenotype in question. For example, if there is a personal or family history of thoracic aortic dissection, genetic testing using a panel of genes causing hereditary aortopathies would be the most rational course of action.
HDCT Diagnosis in Children and Adolescents
Many of the clinical findings associated with HDCTs can affect children and adolescents as well as adults, depending on the specific diagnosis and genetic basis. Manifestations of MFS, for example, can become apparent at any time between infancy and adulthood. Early-onset MFS is a very rare subtype of MFS that can present in the antenatal, neonatal, or infancy period (Abdel-Massih et al., 2002; Ardhanari et al., 2019). Likewise, some
of the rare forms of EDS, such as the arthrochalasia type, present in early childhood (Byers et al., 1997). Hormonal influences that include the onset of puberty have been shown to influence hEDS/HSD; in a survey of women with hEDS/HSD, many reported the appearance or worsening of EDS/HSD symptoms with the onset of puberty (Blagowidow, 2021).
Delayed and Mistaken Diagnosis
Diagnosis of some HDCTs is difficult, potentially leading to delayed or mistaken diagnoses. EDS/HSD diagnosis, for example, is often delayed (mean of 14 years and as long as 28 years) (EURORDIS, 2009, p. 136). Multiple factors may contribute to such delays, including the multisystem, complex, and phenotypically variable nature of the disorders, which leads to a broadly “positive review of systems” such that medical providers are skeptical about patients’ experience of the disorders (Clark, 2021; Halverson et al., 2021). Indeed, patients report experiencing denial among health care providers, as well as family members, as to the reality of their lived experience of certain manifestations of the disorders, such as pain, fatigue, and mild cognitive impairment, sometimes described as “brain fog,” as well as limited treatment options (Clark, 2021; Langhinrichsen-Rohling et al., 2021; Palomo-Toucedo et al., 2020). Providers may mischaracterize the “subjective” experience of some gastrointestinal, neurological, and psychological symptoms as “functional” or “psychogenic” when they are in fact real and quite common neurobiological manifestations of the disorders (Barnum, 2014; Fikree et al., 2017). Research also has documented limited knowledge about HDCTs among health care providers, who often profess a lack of experience with the disorders and discomfort with diagnosing them (Schubart et al., 2021). In general, moreover, access to comprehensive, multidisciplinary care teams with expertise in HDCTs is lacking in terms of both geography and appropriate education (Halverson et al., 2021; Mittal et al., 2021). Finally, many health care professionals have inaccurate expectations that a diagnostic genetic test will be readily available for every HDCT, despite the fact that no such test is available for hEDS or any HSD (Bennett et al., 2021; Halverson et al., 2021).
Effects of Delayed or Mistaken Diagnosis
The long diagnostic odyssey experienced by many patients is often a source of distress and unnecessary hardship (Palomo-Toucedo et al., 2020). That experience may include inappropriate medical interventions and potential iatrogenic harm (EURORDIS, 2009, p. 137). Failure to diagnose an underlying HDCT may lead to inaccurate assessment of the risks and benefits associated with medical procedures, including such routine procedures
as endoscopy or intubation, as well as reduced access to reasonable accommodations for the patient’s condition at work or school.
Family stress and dysfunction may result from family members’ failure to give credence to the symptoms reported by the patient (Halverson et al., 2021). Families may be subjected to stress associated with unexplained and repeated injuries and bruising, including inappropriate suspicion of child abuse (Castori, 2015). Patients report inappropriate assessments and inaccurate diagnoses, and many develop a mistrust of health care providers and negative expectations for future health care encounters, which may lead them to avoid further medical consultations (Halverson et al., 2021; Langhinrichsen-Rohling et al., 2021).
In the case of some of the HDCTs, such as MFS, LDS, and vascular EDS, failure to make the correct diagnosis may be life-threatening. Lack of monitoring for aortic root enlargement in MFS or LDS, for example, may lead to aortic dissection and death, while failure to recognize a characteristic phenotype (e.g., of MFS) in a patient presenting with chest pain may result in delayed diagnosis of aortic dissection, with potentially catastrophic results (Asouhidou and Asteri, 2009; Jarmulowicz and Phillips, 2001; Lovatt et al., 2022).
CLINICAL COURSE
HDCTs are lifelong disorders for which no curative treatments currently exist. Management involves supportive care, treatment of associated secondary impairments, and preventive measures to mitigate or prevent problems that may occur or worsen over time. The fact that HDCTs can manifest in a multitude of physical and mental secondary impairments in virtually any organ system can make the disorders difficult to recognize and manage. In addition, the clinical course of individuals with an HDCT is highly variable. Such variation relates not only to the disease-specific manifestations of each unique syndrome, as discussed above, but also to each individual and that person’s comorbidities and/or underlying conditions. Nevertheless, the natural history of HDCTs as a general group demonstrates several commonalities, including the disorders’ multisystem nature and potential secondary impairments.
Multiple connective tissues can work synchronously to form such structures as joints, where two bones make contact. Hinge joints, such as elbows, which allow for motion, are composed of bone, muscles, synovium, cartilage, and ligaments. These structures and connective tissues are often affected in HDCTs, making joint abnormalities common in most of the disorders. Joint laxity and dislocations are seen in MFS, OI, and EDS, as well as HSD. Joint contractures are seen in CCA, and early-onset degenerative osteoarthritis occurs with many types of EDS and skeletal dysplasia
phenotypes (e.g., Stickler syndrome), illustrating how abnormally organized connective tissues can contribute to organ system dysfunction.
Instability of the joint between head and neck may lead to profound neurologic complications (Henderson et al., 2017). Other potential cranial and spinal neurologic complications are summarized by Debette and Germaine (2014) and Henderson and colleagues (2017). Dysautonomia—failure of the autonomic nervous system to balance properly between the “flight or fight” and “rest and digest” functions—is common in many of the HDCTs (Roma et al., 2018). Increasingly, dysfunction of the mast cells, which are first responders for the immune system, is recognized in several forms of EDS, as well as LDS (Brock et al., 2021). Gastrointestinal issues include motility and barrier dysfunction (i.e., altered intestinal permeability) (Alomari et al., 2020; Fikree et al., 2017; Wong et al., 2022). Mast cell activation and dysautonomia may play a role in gastrointestinal symptoms experienced by some individuals with EDS (Alomari et al., 2020; Wong et al., 2022). In some cases, gastrointestinal complications may lead to malabsorption and nutritional deficiencies (Beckers et al., 2017; Fikree et al., 2017; Wang et al., 2021). Any one or combination of these secondary impairments can contribute to the chronic pain and fatigue experienced by many individuals with HDCTs (Bowen et al., 2017; Hakim et al., 2017; Johansen et al., 2020; Speed et al., 2017; Syx et al., 2017; Tinkle et al., 2017). Mental manifestations (not only reactive) may also contribute to patients’ experience of pain and fatigue. Anxiety, phobias, and depression in particular are very common, may affect the quality of life of individuals with these disorders, and therefore must be properly diagnosed and addressed (Bulbena et al., 2017).
The severity of an HDCT cannot be measured by a single genetic or laboratory test. Rather, the severity of the disorder in an individual is determined by the severity of the person’s physical and mental secondary impairments, which may be measurable with existing clinical and function testing. Severity is also driven by the combined effects of multiple impairments, as well as the frequency, severity, and predictability of their fluctuations. The impact on the individual in terms of functional limitations and restrictions results from the combined effects of the multiple impairments in different body systems, which may be severe collectively even if they are individually graded as “less severe.” This interplay among impairments and the multisystem nature of HDCTs can pose a major difficulty for the global assessment of disease severity.
The functional impairment in HDCTs is also linked to limitations and necessary restrictions due to vulnerability to specific environmental factors and other stressors. For many HDCTs, a patient’s disease severity, manifestations, and clinical course may be adversely affected by specific environmental factors. In addition, physical and mental demands related to school or work may precipitate or exacerbate secondary impairments,
perhaps further limiting activities and restricting participation. Beyond diagnosis of a specific HDCT, informed clinicians can help their patients substantially by being attuned to the many physical and mental secondary impairments with which these patients present. Recognizing the presence of these conditions and taking action to address them, even in the absence of confirmatory genetic testing, can mitigate their effects on the functional status and quality of life of individuals with HDCTs.
DISEASE STATE MANAGEMENT
Central to minimizing the impact of HDCTs on function is a coordinated multidisciplinary management strategy (Miklovic and Sieg, 2021; Mittal et al., 2021). Medical management needs to include a complete body-system review and assessment and periodic monitoring for areas of vulnerability. Functional management needs to address physical vulnerabilities and may require lifelong rehabilitation services. Psychosocial support of patients and families is also important. As noted previously, psychiatric manifestations, including anxiety and depression, are frequently seen in these conditions, as is the case in patients with other chronic conditions. Simple screening tools for depression and anxiety are available for use in the primary care setting. If these conditions are found to be present, a comprehensive mental health evaluation is important, and management may require ongoing assessment and treatment.
High-quality care for individuals with HDCTs relies on effective coordination among a team of providers across a broad range of disciplines (Miklovic and Sieg, 2021; Mittal et al., 2021). As discussed above, access to comprehensive, multidisciplinary care for the diagnosis and management of HDCTs can be limited by geography and other factors, including the availability of care teams with expertise in the disorders (Halverson et al., 2021; Mittal et al., 2021). In addition, access to multidisciplinary teams and relevant specialists is limited or nonexistent in rural areas; even many university centers lack multidisciplinary teams with expertise in HDCTs (Mittal et al., 2021). Education about HDCTs, including their multisystem physical and mental manifestations, diagnosis, and management, is important for all health care providers to increase recognition and earlier diagnosis of the disorders, as well as to improve coordination of their management (Miklovic and Sieg, 2021; Mittal et al., 2021; Schubart et al., 2021). With appropriate education, a variety of clinicians, including, for example, physicians, nurses, psychologists, neuropsychologists, rehabilitation specialists (e.g., physiatrists; physical, occupational, and speech therapists), nutritionists, and others, should be able to recognize HDCTs and direct affected individuals to the appropriate providers for management. Individuals with HDCTs and relevant support groups provide valuable
education regarding the manifestations and lived experience of the disorders (Bloom et al., 2021). Education for patients and families is important to help them understand the multisystem nature of HDCTs and the options available for managing their disorder despite the lack of curative treatment. Information about lifestyle modifications; adherence to physical therapy, medication, and other medical interventions; and symptoms for which to seek immediate medical care can help reduce morbidity (Miklovic and Sieg, 2021). Increased recognition of the breadth and scope of HDCTs by health care professional education programs, professional organizations, and publishers of quality biomedical research is needed.
FINDINGS AND CONCLUSIONS
Findings
2-1. Heritable disorders of connective tissue (HDCTs) are a heterogeneous group of inherited disorders that affect connective tissues in organ systems throughout the body.
2-2. Connective tissues are an integral component of every organ system and play a crucial role in the function of those systems. Hence, the physical and mental secondary impairments associated with HDCTs, which may develop and potentially progress or wax and wane over time, manifest throughout the body and affect functioning in every body system.
2-3. HDCTs can be difficult to diagnose, and the true prevalence of many of these disorders is unknown.
2-4. Because HDCTs can cause a wide variety of physical and mental secondary impairments involving multiple organ systems, affected individuals often are referred to a succession of different specialists, resulting in delayed diagnosis of the underlying HDCT.
2-5. HDCTs are diagnosed through a combination of clinical findings and established clinical criteria, followed by confirmatory molecular genetic testing when specific genes have been identified for the suspected disorder.
2-6. HDCTs are lifelong disorders for which no curative treatments currently exist. Management involves supportive care, treatment of associated secondary impairments, and preventive measures to mitigate or prevent problems that may occur or worsen over time.
2-7. Individuals with HDCTs often experience difficulty with obtaining appropriate and integrated multidisciplinary care to address the wide range of physical and mental impairments associated with these disorders.
2-8. Access to comprehensive, multidisciplinary care for the diagnosis and management of HDCTs can be limited by geography and other factors, including the availability of care teams with expertise in the disorders.
2-9. The clinical course of HDCTs is highly variable and can be impacted not only by the disease-specific manifestations of each unique syndrome, but also by individuals’ physical and mental secondary impairments, as well as environmental factors and physical and psychological demands.
2-10. The severity of HDCTs is linked to the severity of affected individuals’ physical and mental secondary impairments, including the combined effects of multiple impairments, as well as the frequency, severity, and predictability of their fluctuations.
Conclusions
2-1. Consideration of a diagnosis of an HDCT is warranted for individuals who present with previously undiagnosed complex multisystem disorders.
2-2. Early diagnosis of HDCTs is important to reduce physical injury, reduce psychological harm to affected individuals and their family members, and prevent the risks associated with inappropriate medical care.
2-3. Appropriate multisystem assessments are important at the time of HDCT diagnosis and at intervals across a person’s life.
2-4. Appropriate multidisciplinary understanding and management of HDCTs can reduce the frequency and severity of their manifestations and resulting functional limitations.
REFERENCES
Abdel-Massih, T., A. Goldenberg, P. Vouhé, F. Iserin, P. Acar, E. Villain, G. Agnoletti, D. Sidi, and D. Bonnet. 2002. Marfan syndrome in the newborn and infants less than 4 months: A series of 9 patients. Archives des Maladies du Coeur et des Vaisseaux 95(5):469-472.
Abdo, H., L. Calvo-Enrique, J. M. Lopez, J. Song, M. D. Zhang, D. Usoskin, A. El Manira, I. Adameyko, J. Hjerling-Leffler, and P. Ernfors. 2019. Specialized cutaneous schwann cells initiate pain sensation. Science 365(6454):695-699. https://doi.org/10.1126/science.aax6452.
Alomari, M., A. Hitawala, P. Chadalavada, F. Covut, L. A. Momani, S. Khazaaleh, F. Gosai, S. A. Ashi, A. Abushahin, and A. Schneider. 2020. Prevalence and predictors of gastrointestinal dysmotility in patients with hypermobile Ehlers-Danlos syndrome: A tertiary care center experience. Cureus 12(4):e7881. https://doi.org/10.7759/cureus.7881.
Ardhanari, M., D. Barbouth, and S. Swaminathan. 2019. Early-onset Marfan syndrome: A case series. Journal of Pediatric Genetics 8(2):86-90. https://doi.org/10.1055/s-0038-1675338.
Arnaud, P., O. Milleron, N. Hanna, J. Ropers, N. Ould Ouali, A. Affoune, M. Langeois, L. Eliahou, F. Arnoult, P. Renard, M. Michelon-Jouneaux, M. Cotillon, L. Gouya, C. Boileau, and G. Jondeau. 2021. Clinical relevance of genotype–phenotype correlations beyond vascular events in a cohort study of 1500 Marfan syndrome patients with FBN1 pathogenic variants. Genetics in Medicine 23(7):1296-1304. https://doi.org/10.1038/s41436-021-01132-x.
Asahara, H., M. Inui, and M. K. Lotz. 2017. Tendons and ligaments: Connecting developmental biology to musculoskeletal disease pathogenesis. Journal of Bone and Mineral Research 32(9):1773-1782. https://doi.org/10.1002/jbmr.3199.
Asouhidou, I., and T. Asteri. 2009. Acute aortic dissection: Be aware of misdiagnosis. BMC Research Notes 2:25. https://doi.org/10.1186/1756-0500-2-25.
Barallobre-Barreiro, J., B. Loeys, M. Mayr, M. Rienks, A. Verstraeten, and J. C. Kovacic. 2020. Extracellular matrix in vascular disease, part 2/4: JACC Focus Seminar. Journal of the American College of Cardiology 75(17):2189-2203. https://doi.org/10.1016/j.jacc.2020.03.018.
Barnum, R. 2014. Problems with diagnosing conversion disorder in response to variable and unusual symptoms. Adolescent Health, Medicine and Therapeutics 5:67-71. https://doi.org/10.2147/ahmt.S57486.
Beckers, A. B., D. Keszthelyi, A. Fikree, L. Vork, A. Masclee, A. D. Farmer, and Q. Aziz. 2017. Gastrointestinal disorders in joint hypermobility syndrome/Ehlers-Danlos syndrome hypermobility type: A review for the gastroenterologist. Neurogastroenterology and Motility 29(8):e13013. https://doi.org/10.1111/nmo.13013.
Bennett, S. E., N. Walsh, T. Moss, and S. Palmer. 2021. Understanding the psychosocial impact of joint hypermobility syndrome and Ehlers-Danlos syndrome hypermobility type: A qualitative interview study. Disability and Rehabilitation 43(6):795-804. https://doi.org/10.1080/09638288.2019.1641848.
Blagowidow, N. 2021. Obstetrics and gynecology in Ehlers-Danlos syndrome: A brief review and update. American Journal of Medical Genetics Part C: Seminars in Medical Genetics 187(4):593-598. https://doi.org/10.1002/ajmg.c.31945.
Bloom, L., J. Schubart, R. Bascom, A. Hakim, and C. A. Francomano. 2021. The power of patient-led global collaboration. American Journal of Medical Genetics Part C: Seminars in Medical Genetics 187(4):425-428. https://doi.org/10.1002/ajmg.c.31942.
Bowen, J. M., G. J. Sobey, N. P. Burrows, M. Colombi, M. E. Lavallee, F. Malfait, and C. A. Francomano. 2017. Ehlers-Danlos syndrome, classical type. American Journal of Medical Genetics Part C: Seminars in Medical Genetics 175(1):27-39. https://doi.org/10.1002/ajmg.c.31548.
Brock, I., W. Prendergast, and A. Maitland. 2021. Mast cell activation disease and immunoglobulin deficiency in patients with hypermobile Ehlers-Danlos syndrome/hypermobility spectrum disorder. American Journal of Medical Genetics. Part C: Seminars in Medical Genetics 187(4):473-481. https://doi.org/10.1002/ajmg.c.31940.
Bulbena, A., C. Baeza-Velasco, A. Bulbena-Cabré, G. Pailhez, H. Critchley, P. Chopra, N. Mallorquí-Bagué, C. Frank, and S. Porges. 2017. Psychiatric and psychological aspects in the Ehlers–Danlos syndromes. American Journal of Medical Genetics Part C: Seminars in Medical Genetics 175(1):237-245. https://doi.org/10.1002/ajmg.c.31544.
Byers, P. H. 2019. Vascular Ehlers-Danlos syndrome. In GeneReviews® [Internet], edited by M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. W. Gripp, G. M. Mirzaa, and A. Amemiya. Seattle, WA: University of Washington; 1993-2022. https://www.ncbi.nlm.nih.gov/books/NBK1494/.
Byers, P. H., M. Duvic, M. Atkinson, M. Robinow, L. T. Smith, S. M. Krane, M. T. Greally, M. Ludman, R. Matalon, S. Pauker, D. Quanbeck, and U. Schwarze. 1997. Ehlers-Danlos syndrome type VIIA and VIIB result from splice-junction mutations or genomic deletions that involve exon 6 in the COL1A1 and COL1A2 genes of type I collagen. American Journal of Medical Genetics 72(1):94-105. https://doi.org/10.1002/(sici)1096-8628(19971003)72:1<94::aid-ajmg20>3.0.co;2-o.
Castori, M. 2015. Ehlers-Danlos syndrome(s) mimicking child abuse: Is there an impact on clinical practice? American Journal of Medical Genetics Part C: Seminars in Medical Genetics 169(4):289-292. https://doi.org/10.1002/ajmg.c.31460.
Clark, S. 2021. Help me trust you after my misdiagnosis. BMJ 373:n1175. https://doi.org/10.1136/bmj.n1175.
Connizzo, B. K., S. M. Yannascoli, and L. J. Soslowsky. 2013. Structure-function relationships of postnatal tendon development: A parallel to healing. Matrix Biology 32(2):106-116. https://doi.org/10.1016/j.matbio.2013.01.007.
Dawidowicz, J., S. Szotek, N. Matysiak, Ł. Mielańczyk, and K. Maksymowicz. 2015. Electron microscopy of human fascia lata: Focus on telocytes. Journal of Cellular and Molecular Medicine 19(10):2500-2506. https://doi.org/10.1111/jcmm.12665.
Debette, S., and D. P. Germain. 2014. Chapter 37—Neurologic manifestations of inherited disorders of connective tissue. Handbook of Clinical Neurology 119:565-576. https://doi.org/10.1016/b978-0-7020-4086-3.00037-0.
del Monte-Nieto, G., J. W. Fischer, D. J. Gorski, R. P. Harvey, and J. C. Kovacic. 2020. Basic biology of extracellular matrix in the cardiovascular system, part 1/4: JACC Focus Seminar. Journal of the American College of Cardiology 75(17):2169-2188. https://doi.org/10.1016/j.jacc.2020.03.024.
EURORDIS. 2009. The voice of 12,000 patients. https://www.eurordis.org/publication/voice-12000-patients.
Fikree, A., G. Chelimsky, H. Collins, K. Kovacic, and Q. Aziz. 2017. Gastrointestinal involvement in the Ehlers-Danlos syndromes. American Journal of Medical Genetics Part C: Seminars in Medical Genetics 175(1):181-187. https://doi.org/10.1002/ajmg.c.31546.
Follis, R. H., Jr. 1952. Osteogenesis imperfecta congenita: A connective tissue diathesis. The Journal of Pediatrics 41(6):713-721. https://doi.org/10.1016/s0022-3476(52)80292-6.
Follis, R. H., Jr. 1953a. Histochemical studies on cartilage and bone. III. Osteogenesis imperfecta. Bulletin of the Johns Hopkins Hospital 93(6):386-399.
Follis, R. H., Jr. 1953b. Maldevelopment of the corium in the osteogenesis imperfecta syndrome. Bulletin of the Johns Hopkins Hospital 93(4):225-233.
Gillies, A. R., and R. L. Lieber. 2011. Structure and function of the skeletal muscle extracellular matrix. Muscle & Nerve 44(3):318-331. https://doi.org/10.1002/mus.22094.
Glass, D., A. Viñuela, M. N. Davies, A. Ramasamy, L. Parts, D. Knowles, A. A. Brown, Å. K. Hedman, K. S. Small, A. Buil, E. Grundberg, A. C. Nica, P. Di Meglio, F. O. Nestle, M. Ryten, the UK Brain Expression consortium, the MUther consortium, R. Durbin, M. I. McCarthy, P. Deloukas, E. T. Dermitzakis, M. E. Weale, V. Bataille, and T. D. Spector, 2013. Gene expression changes with age in skin, adipose tissue, blood and brain. Genome Biology 14(7):R75. https://doi.org/10.1186/gb-2013-14-7-r75.
Hakim, A., I. De Wandele, C. O’Callaghan, A. Pocinki, and P. Rowe. 2017. Chronic fatigue in Ehlers-Danlos syndrome-hypermobile type. American Journal of Medical Genetics Part C: Seminars in Medical Genetics 175(1):175-180. https://doi.org/10.1002/ajmg.c.31542.
Halata, Z. 1977. The ultrastructure of the sensory nerve endings in the articular capsule of the knee joint of the domestic cat (Ruffini corpuscles and Pacinian corpuscles). Journal of Anatomy 124(Pt 3):717-729. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1234668/.
Halverson, C. M. E., E. W. Clayton, A. Garcia Sierra, and C. Francomano. 2021. Patients with Ehlers–Danlos syndrome on the diagnostic odyssey: Rethinking complexity and difficulty as a hero’s journey. American Journal of Medical Genetics Part C: Seminars in Medical Genetics 187(4):416-424. https://doi.org/10.1002/ajmg.c.31935.
Hayakawa, M., M. Kobayashi, and T. Hoshino. 1990. Microfibrils: A constitutive component of reticular fibers in the mouse lymph node. Cell and Tissue Research 262(1):199-201. https://doi.org/10.1007/bf00327763.
Henderson, F. C., Sr., C. Austin, E. Benzel, P. Bolognese, R. Ellenbogen, C. A. Francomano, C. Ireton, P. Klinge, M. Koby, D. Long, S. Patel, E. L. Singman, and N. C. Voermans. 2017. Neurological and spinal manifestations of the Ehlers-Danlos syndromes. American Journal of Medical Genetics. Part C: Seminars in Medical Genetics 175(1):195-211. https://doi.org/10.1002/ajmg.c.31549.
Işıldak, U., M. Somel, J. M. Thornton, and H. M. Dönertaş. 2020. Temporal changes in the gene expression heterogeneity during brain development and aging. Scientific Reports 10(1):4080. https://doi.org/10.1038/s41598-020-60998-0.
Jarmulowicz, M., and W. G. Phillips. 2001. Vascular Ehlers-Danlos syndrome undiagnosed during life. Journal of the Royal Society of Medicine 94(1):28-30. https://doi.org/10.1177/014107680109400108.
Jensen, S. A., and P. A. Handford. 2016. New insights into the structure, assembly and biological roles of 10–12 nm connective tissue microfibrils from fibrillin-1 studies. Biochemical Journal 473(7):827-838. https://doi.org/10.1042/BJ20151108.
Johansen, H., G. Velvin, and I. Lidal. 2020. Adults with Loeys–Dietz syndrome and vascular Ehlers–Danlos syndrome: A cross-sectional study of health burden perspectives. American Journal of Medical Genetics Part A 182(1):137-145. https://doi.org/10.1002/ajmg.a.61396.
Judge, D. P., and H. C. Dietz. 2005. Marfan’s syndrome. Lancet 366(9501):1965-1976. https://doi.org/10.1016/s0140-6736(05)67789-6.
Kannus, P. 2000. Structure of the tendon connective tissue. Scandinavian Journal of Medicine & Science in Sports 10(6):312-320. https://doi.org/10.1034/j.1600-0838.2000.010006312.x.
Knuutinen, A., N. Kokkonen, J. Risteli, K. Vähäkangas, M. Kallioinen, T. Salo, T. Sorsa, and A. Oikarinen. 2002. Smoking affects collagen synthesis and extracellular matrix turnover in human skin. British Journal of Dermatology 146(4):588-594. https://doi.org/10.1046/j.1365-2133.2002.04694.x.
Krakower, C. 1972. The cells and tissues of the immune system: Structure, functions, interactions. JAMA 220(10):1366. https://doi.org/10.1001/jama.1972.03200100076031.
Langhinrichsen-Rohling, J., C. L. Lewis, S. McCabe, E. C. Lathan, G. A. Agnew, C. N. Selwyn, and M. E. Gigler. 2021. They’ve been BITTEN: Reports of institutional and provider betrayal and links with Ehlers-Danlos syndrome patients’ current symptoms, unmet needs and healthcare expectations. Therapeutic Advances in Rare Disease 2:263300402110220. https://dx.doi.org/10.1177/26330040211022033.
Liu, T., T. H. McCalmont, I. J. Frieden, M. L. Williams, M. K. Connolly, and A. E. Gilliam. 2008. The stiff skin syndrome: Case series, differential diagnosis of the stiff skin phenotype, and review of the literature. Archives of Dermatology 144(10):1351-1359. https://doi.org/10.1001/archderm.144.10.1351.
Loeys, B. L., and H. C. Dietz. 2018. Loeys-Dietz syndrome. In GeneReviews® [Internet], edited by M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. W. Gripp, G. M. Mirzaa and A. Amemiya. Seattle, WA: University of Washington; 1993-2022. https://www.ncbi.nlm.nih.gov/books/NBK1133/.
Loeys, B. L., E. E. Gerber, D. Riegert-Johnson, S. Iqbal, P. Whiteman, V. McConnell, C. R. Chillakuri, D. Macaya, P. J. Coucke, A. De Paepe, D. P. Judge, F. Wigley, E. C. Davis, H. J. Mardon, P. Handford, D. R. Keene, L. Y. Sakai, and H. C. Dietz. 2010. Mutations in fibrillin-1 cause congenital scleroderma: Stiff skin syndrome. Science Translational Medicine 2(23):23ra20. https://doi.org/10.1126/scitranslmed.3000488.
Lovatt, S., C. W. Wong, K. Schwarz, J. A. Borovac, T. Lo, M. Gunning, T. Phan, A. Patwala, D. Barker, C. D. Mallen, and C. S. Kwok. 2022. Misdiagnosis of aortic dissection: A systematic review of the literature. American Journal of Emergency Medicine 53:16-22. https://doi.org/10.1016/j.ajem.2021.11.047.
McKusick, V. A. 1955. The cardiovascular aspects of Marfan’s syndrome: A heritable disorder of connective tissue. Circulation 11(3):321-342. https://doi.org/10.1161/01.cir.11.3.321.
Miklovic, T., and V. Sieg. 2021. Ehlers Danlos syndrome. In StatPearls [Internet]. Treasure Island, FL: StatPearls Publishing. https://www.ncbi.nlm.nih.gov/books/NBK549814/.
Milewicz, D. M., A. C. Braverman, J. De Backer, S. A. Morris, C. Boileau, I. H. Maumenee, G. Jondeau, A. Evangelista, and R. E. Pyeritz. 2021. Marfan syndrome. Nature Reviews: Disease Primers 7(1):64. https://doi.org/10.1038/s41572-021-00298-7.
Mittal, N., D. S. Mina, L. McGillis, A. Weinrib, P. M. Slepian, M. Rachinsky, S. Buryk-Iggers, C. Laflamme, L. Lopez-Hernandez, L. Hussey, J. Katz, L. McLean, D. Rozenberg, L. Liu, Y. Tse, C. Parker, A. Adler, G. Charames, R. Bleakney, C. Veillette, C. J. Nielson, S. Tavares, S. Varriano, J. Guzman, H. Faghfoury, and H. Clarke. 2021. The GoodHope Ehlers Danlos Syndrome Clinic: Development and implementation of the first interdisciplinary program for multi-system issues in connective tissue disorders at the Toronto General Hospital. Orphanet Journal of Rare Diseases 16(1):357. https://doi.org/10.1186/s13023-021-01962-7.
Mortier, G. R., D. H. Cohn, V. Cormier-Daire, C. Hall, D. Krakow, S. Mundlos, G. Nishimura, S. Robertson, L. Sangiorgi, R. Savarirayan, D. Sillence, A. Superti-Furga, S. Unger, and M. L. Warman. 2019. Nosology and classification of genetic skeletal disorders: 2019 revision. American Journal of Medical Genetics Part A 179(12):2393-2419. https://doi.org/10.1002/ajmg.a.61366.
Nezwek, T. A., and M. Varacallo. 2021. Physiology, connective tissue. In StatPearls [Internet]. Treasure Island, FL: StatPearls Publishing. https://www.ncbi.nlm.nih.gov/books/NBK542226/.
NORD (National Organization for Rare Disorders). 2021. Rare disease database: Marfan syndrome. https://rarediseases.org/rare-diseases/marfan-syndrome/ (accessed February 9, 2022).
Omelyanenko, N. P., and L. I. Slutsky. 2014. Connective tissue: Histophysiology, biochemistry, molecular biology. 1st ed. Boca Raton, FL: CRC Press. https://doi.org/10.1201/b16297
Orphanet. 2022a. Dermatosparaxis Ehlers-Danlos syndrome. https://www.orpha.net/consor4.01/www/cgi-bin/Disease_Search.php?lng=EN&data_id=4045&Disease_Disease_Search_diseaseGroup=Dermatosparaxis-EDS&Disease_Disease_Search_diseaseType=Pat&Disease(s)/group%20of%20diseases=Dermatosparaxis-Ehlers-Danlos-syndrome&title=Dermatosparaxis%20Ehlers-Danlos%20syndrome&search=Disease_Search_Simple (accessed February 16, 2022).
Orphanet. 2022b. Musculocontractural Ehlers-Danlos syndrome. https://www.orpha.net/consor4.01/www/cgi-bin/Disease_Search.php?lng=EN&data_id=3480&Disease_Disease_Search_diseaseGroup=Musculocontractural-EDS&Disease_Disease_Search_diseaseType=Pat&Disease(s)/group%20of%20diseases=Musculocontractural-Ehlers-Danlos-syndrome&title=Musculocontractural%20Ehlers-Danlos%20syndrome&search=Disease_Search_Simple (accessed February 16, 2022).
Paiva, K. B. S., and J. M. Granjeiro. 2017. Chapter six—Matrix metalloproteinases in bone resorption, remodeling, and repair. Progress in Molecular Biology and Translational Science 148:203-303. https://doi.org/10.1016/bs.pmbts.2017.05.001.
Palomo-Toucedo, I. C., F. Leon-Larios, M. Reina-Bueno, M. D. C. Vázquez-Bautista, P. V. Munuera-Martínez, and G. Domínguez-Maldonado. 2020. Psychosocial influence of Ehlers-Danlos syndrome in daily life of patients: A qualitative study. International Journal of Environmental Research and Public Health 17(17). https://doi.org/10.3390/ijerph17176425.
Pyeritz, R. E. 2000. Ehlers-Danlos syndromes. In Cecil textbook of medicine. 21st ed. Vol. 1, edited by L. Goldman and J. C. Bennett. Philadelphia: W.B. Saunders. Pp. 1119-1120.
Ricard-Blum, S. 2011. The collagen family. Cold Spring Harbor Perspectives in Biology 3(1):a004978. https://doi.org/10.1101/cshperspect.a004978.
Richards, S., N. Aziz, S. Bale, D. Bick, S. Das, J. Gastier-Foster, W. W. Grody, M. Hegde, E. Lyon, E. Spector, K. Voelkerding, and H. L. Rehm. 2015. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine 17(5):405-423. https://doi.org/10.1038/gim.2015.30.
Roma, M., C. L. Marden, I. De Wandele, C. A. Francomano, and P. C. Rowe. 2018. Postural tachycardia syndrome and other forms of orthostatic intolerance in Ehlers-Danlos syndrome. Autonomic Neuroscience 215:89-96. https://doi.org/10.1016/j.autneu.2018.02.006.
Sakai, L. Y., and D. R. Keene. 2019. Fibrillin protein pleiotropy: Acromelic dysplasias. Matrix Biology 80:6-13. https://doi.org/10.1016/j.matbio.2018.09.005.
Savarirayan, R., V. Bompadre, M. B. Bober, T.-J. Cho, M. J. Goldberg, J. Hoover-Fong, M. Irving, S. E. Kamps, W. G. Mackenzie, C. Raggio, S. S. Spencer, and K. K. White. 2019. Best practice guidelines regarding diagnosis and management of patients with type II collagen disorders. Genetics in Medicine 21(9):2070-2080. https://doi.org/10.1038/s41436-019-0446-9.
Schubart, J. R., R. Bascom, C. A. Francomano, L. Bloom, and A. J. Hakim. 2021. Initial description and evaluation of EDS ECHO: An international effort to improve care for people with the Ehlers-Danlos syndromes and hypermobility spectrum disorders. American Journal of Medical Genetics Part C: Seminars in Medical Genetics 187(4):609-615. https://doi.org/10.1002/ajmg.c.31960.
Shukunami, C., A. Takimoto, M. Oro, and Y. Hiraki. 2006. Scleraxis positively regulates the expression of tenomodulin, a differentiation marker of tenocytes. Developmental Biology 298(1):234-247. https://doi.org/10.1016/j.ydbio.2006.06.036.
Sophia Fox, A. J., A. Bedi, and S. A. Rodeo. 2009. The basic science of articular cartilage: Structure, composition, and function. Sports Health 1(6):461-468. https://doi.org/10.1177/1941738109350438.
Speed, T. J., V. A. Mathur, M. Hand, B. Christensen, P. D. Sponseller, K. A. Williams, and C. M. Campbell. 2017. Characterization of pain, disability, and psychological burden in Marfan syndrome. American Journal of Medical Genetics Part A 173(2):315-323. https://doi.org/10.1002/ajmg.a.38051.
Stecco, C., C. Fede, V. Macchi, A. Porzionato, L. Petrelli, C. Biz, R. Stern, and R. De Caro. 2018. The fasciacytes: A new cell devoted to fascial gliding regulation. Clinical Anatomy 31(5):667-676. https://doi.org/10.1002/ca.23072.
Steinmann, B., P. M. Royce, and A. Superti-Furga. 2002. The Ehlers-Danlos syndrome. In Connective tissue and its heritable disorders: Molecular, genetic, and medical aspects. 2nd ed., edited by B. Steinmann and P. M. Royce. New York: Wiley‐Liss, Inc. Pp. 431-524. https://doi.org/10.1002/0471221929.ch9.
Syx, D., I. De Wandele, L. Rombaut, and F. Malfait. 2017. Hypermobility, the Ehlers-Danlos syndromes and chronic pain. Clinical and Experimental Rheumatology 35 Suppl 107(5):116-122.
Tinkle, B., M. Castori, B. Berglund, H. Cohen, R. Grahame, H. Kazkaz, and H. Levy. 2017. Hypermobile Ehlers-Danlos syndrome (a.k.a. Ehlers-Danlos syndrome type III and Ehlers-Danlos syndrome hypermobility type): Clinical description and natural history. American Journal of Medical Genetics Part C: Seminars in Medical Genetics 175(1):48-69. https://doi.org/10.1002/ajmg.c.31538.
Uitto, J. 1979. Biochemistry of the elastic fibers in normal connective tissues and its alterations in diseases. Journal of Investigative Dermatology 72(1):1-10. https://doi.org/10.1111/1523-1747.ep12530093.
University of Leeds. 2002. Oral: Four layers of the G.I. tract. https://www.histology.leeds.ac.uk/digestive/GI_layers.php (accessed July 19, 2022).
van Deemter, M., H. H. Pas, R. Kuijer, R. J. van der Worp, J. M. M. Hooymans, and L. I. Los. 2009. Enzymatic breakdown of type II collagen in the human vitreous. Investigative Ophthalmology & Visual Science 50(10):4552–4560. https://doi.org/10.1167/iovs.08-3125.
Vandenabeele, F., J. Creemers, I. Lambrichts, P. Lippens, and M. Jans. 1997. Encapsulated Ruffini-like endings in human lumbar facet joints. Journal of Anatomy 191(Pt 4):571-583. https://doi.org/10.1046/j.1469-7580.1997.19140571.x.
Wang, X. J., M. Babameto, D. Babovic-Vuksanovic, J. M. Bowen, and M. Camilleri. 2021. Audit of gastrointestinal manifestations in patients with Loeys-Dietz syndrome and vascular Ehlers-Danlos syndrome. Digestive Diseases and Sciences 66(4):1142-1152. https://doi.org/10.1007/s10620-020-06265-8.
Watanabe, T., Y. Hosaka, E. Yamamoto, H. Ueda, P. Tangkawattana, and K. Takehana. 2004. Morphological study of the Golgi tendon organ in equine superficial digital flexor tendon. Okajimas Folia Anatomica Japonica 81(2-3):33-37. https://doi.org/10.2535/ofaj.81.33.
Weatherholt, A. M., R. K. Fuchs, and S. J. Warden. 2012. Specialized connective tissue: Bone, the structural framework of the upper extremity. Journal of Hand Therapy 25(2):123-132. https://doi.org/10.1016/j.jht.2011.08.003.
Wong, S., S. Hasan, C. Parducci, and B. A. Riley. 2022. The gastrointestinal effects amongst Ehlers-Danlos syndrome, mast cell activation syndrome and postural orthostatic tachycardia syndrome. AIMS Allergy and Immunology 6(2):19-24. https://www.aimspress.com/article/doi/10.3934/Allergy.2022004.
This page intentionally left blank.






















