
4
Atmospheric Transport and Chemical Transformations
Wildland-urban interface (WUI) fires can have substantial negative impacts on human health, visibility, and quality of life, not only in the vicinity of the fire, but also hundreds of kilometers downwind. For example, even 240 km downwind of the Camp Fire, in the Bay Area of northern California, daily fine particulate matter (PM2.5) concentrations remained elevated (70–200 μg/m3) for over 2 weeks, well above the National Ambient Air Quality Standard of 35 μg/m3 (Rooney et al., 2020). The population experiencing this exposure was in excess of seven million people. Thus, this WUI fire resulted in substantial adverse particulate matter exposures to large populations beyond the immediate zone of the fire. Smoke from major fires sometimes affects air quality and visibility on a continental scale, and seasonally averaged summertime contributions from wildland fire smoke PM2.5 can be comparable to contributions from non-smoke PM2.5 in some US regions (e.g., in the Pacific Northwest; O’Dell et al., 2019).
Finding: A large number of people are exposed to WUI fire emissions over broad geographic areas.
While national monitoring networks provide data for assessing exposures of routinely monitored air pollutants, such as fine particulate matter, the gas- and particle-phase smoke composition specifically associated with WUI fires is not well understood. WUI smoke composition differs from wildland fire smoke because of direct emissions from the combustion and volatilization of human-made materials and the chemical interaction of these emissions with wildland fire emissions (see Chapter 3). Atmospheric chemistry and physical changes (e.g., dilution; phase changes; changes in partitioning of compounds between the gas phase, particles, and cloud droplets; deposition), which are dynamic on timescales of minutes to days, lead to dramatic changes in pollutant composition downwind of fires. However, the impact of WUI emissions and chemistry on regional exposures is not well understood. This chapter explores this impact, emphasizing atmospheric chemistry that informs inhalation exposures and the resulting health effects associated with emissions from WUI fires.
It is important to recognize that, in addition to chemical processes, the physical processes of wet and dry atmospheric deposition can also be a source of contaminated water and soil and thus impact exposures through ingestion. Organic nitrogen (Yu et al., 2020), isocyanic acid (Roberts and Liu, 2019; Roberts et al., 2011) and per- and polyfluoroalkyl substances (PFASs; Pike et al., 2021; Shimizu et al., 2021) are examples of water-soluble compounds found in emissions from burning biomass or in emissions from burning human-made materials. Wet deposition of water-soluble compounds can contribute to water and/or soil contamination and exposure through WUI fire–associated wet deposition. The topic of water and soil contamination is examined in Chapter 5.
Finally, while this chapter will not cover visibility, it is worth noting that the impacts of WUI fires on visibility can be substantial and can impact transportation safety, property values, mental health, and quality of life.
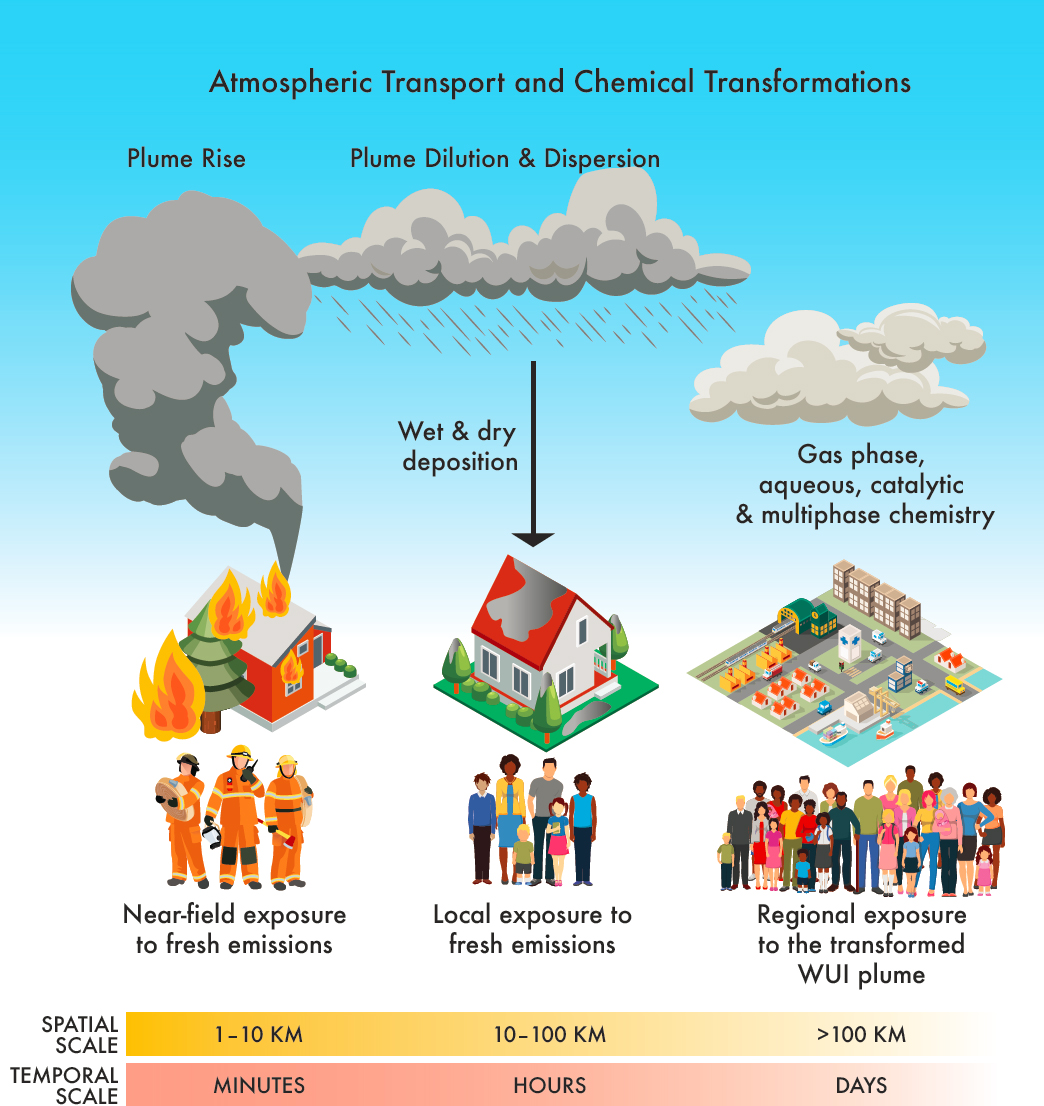
In describing chemical processes that affect the concentrations and composition of air pollutants from WUI fires, the committee defines four zones beyond the fire: the near-field, local, regional, and continental scales. Box 4-1 defines these spatial scales and other key terminology used in the chapter.
PRIMARY SPECIES WITH TOXIC POTENTIAL DOWNWIND OF WUI FIRES
Table 4-1 lists some of the toxicants expected to be directly emitted from WUI fires (i.e., the primary pollutants) and processes that impact their transport, their partitioning between the gas and particle phases in the atmosphere, their transformation, and their degradation. This list draws from laboratory and field measurements focusing on wildland fires, structure fires, and WUI fires, and information about materials (see also Chapter 3). An estimate of species’ atmospheric lifetimes is provided. This estimate is based on the primary degradation pathways being photolysis, reaction with hydroxyl radical, and reaction with ozone in the gas phase. Light intensities, hydroxyl and other radical concentrations, and ozone concentrations that the toxicants are exposed to will vary considerably, so these lifetime estimates are approximate and relative indicators of the persistence of the individual chemical species. The possibility that other loss processes dominate in WUI fire plumes (e.g., oxidation by chlorine radical, multiphase chemistry, cloud chemistry) is discussed in the next section, “Atmospheric Transformations.”
ATMOSPHERIC TRANSFORMATIONS
The Downwind Fate of WUI Fire Emissions
A wide range of plume studies including studies of wildland fire plumes provide confidence that WUI smoke concentrations decrease with distance downwind through dilution and surface deposition and undergo chemical and phase changes in the atmosphere (Figure 4-1); however, the influence of various chemical processes on the downwind composition of WUI fire plumes is poorly understood. Exposures (i.e., who is exposed, to what, and at what concentrations) depend most on whether the emissions are lofted above the mixed layer and transported long distances before being downmixed to ground level or are trapped near the surface in the near field. Plume rise (i.e., plume injection height) depends on the overall heat released by the fire, as discussed in Chapter 3, and is a major factor determining transport altitude. Meteorology, including transport altitude—the location of the plume within or above the layer of the atmosphere that is well mixed (mixed layer)—also affects toxicant concentrations, gas-particle partitioning, photochemistry, cloud processing, and multiphase chemistry (e.g., through its influence on dilution, temperature and saturation vapor pressure, solar radiation, aerosol liquid water content, and phase separation) as described for wildland fires by Jaffe et al. (2020). Photochemical oxidation in a wildland fire plume can produce elevated ground-level ozone (Dreessen et al., 2016; Lu et al., 2016) and oxidized aerosol on a regional scale (Garofalo et al., 2019; Junghenn Noyes et al., 2020). This is also expected in WUI fire plumes.
Finding: Atmospheric chemistry transforms emissions with distance from the fire, so that downwind communities are exposed to more oxidized mixtures than near-field communities.
A variety of chemical and physical processes that take place on timescales of minutes to days reduce the concentrations of primary species with toxic potential and can also form secondary species with toxic potential. A distinguishing feature of wildland fires, as an emission source, is the prevalence of incomplete combustion and thermal degradation of oxygenated fuel components, such as cellulose, when compared to most primary emission sources such as automotive exhaust. This results in an abundance of primary oxygenated, water-soluble compounds in wildland fire emissions (e.g., HONO, HNO3, HCl, formaldehyde, glycolaldehyde, acetic acid, phenols, furfurals, isocyanic acid, and amines; Hatch et al., 2015; Friedli et al., 2001; Juncosa Calahorrano et al., 2021; Koss et al., 2018; Roberts et al., 2011; Selimovic et al., 2018; Tomaz et al., 2018). Comprehensive volatile organic compound (VOC) measurements from controlled burns of 18 US wildland fire fuels suggest
VOC emissions from wildland fires are roughly 60 percent oxygenated (Gilman et al., 2015). The atmospheric fate of water-soluble compounds is determined by the competition between the rates of gas-phase photochemical reactions, reactive uptake in clouds and wet aerosols, and wet deposition; understanding the fate of water-soluble compounds in wildland fire plumes is an active area of research.
The impacts of fire emissions from human-made materials and firefighting activities on the chemistry and fate of WUI fire plumes is not well understood. Relative to wildland fire emissions, WUI emissions have increased levels of several types of compounds, including reactive halogenated compounds (HCl, HBr, HF), dioxins, phenols from the degradation of polymers, aldehydes, nitrogen-containing organics such as isocyanates, brominated and fluorinated organics, and metals (see Chapter 3 and Table 4-1). These species and radicals derived from their atmospheric chemistry could alter the rate of chemical transformations in WUI plumes, changing the lifetime of primary species with toxic potential and the formation of secondary species with toxic potential. The abundance, atmospheric dynamics, and fate of WUI-enriched species are of interest for several reasons, as discussed below.
Near-Field Changes in Partitioning with Dilution
At the near-field scale, downwind of the immediate zone of the fire, dilution of emissions with clean air results in near-field smoke concentrations that are substantially lower than concentrations in the immediate zone. It also results in substantial evaporation from primary organic aerosols, shifting some compounds from the particle to the gas phase. This alters the fate of the mixture and the dose inhaled by exposed populations. For example, laboratory studies of a primary diesel combustion aerosol suggest that dilution to ambient conditions results in evaporation of roughly three quarters of the primary organic aerosol (Robinson et al., 2007, Figure 1A).
Several intermediate-volatility organic compounds (IVOCs), which evaporate during dilution, can subsequently undergo gas-phase oxidation over minutes to hours (i.e., at the near-field and local scales), changing the composition and increasing the oxidation state of organics in both the gas and particle phases (Lambe et al., 2012; Robinson et al., 2007). Phenol and guaiacol, formed through the thermal degradation of lignin, are examples of IVOC wildland fire emissions (Alves et al., 2011; Hatch et al., 2018; Yee et al., 2013).
WUI fire emissions also have increased levels of dioxins and synthetic polymer degradation products (see Chapter 3; Reisch, 2018; Ruokojärvi et al., 2000), some of which are IVOCs and could evaporate from emitted particles with near-field dilution. Some PFASs are also IVOCs. While they are only a small portion of the total mass of human-made materials associated with structures, PFASs are ubiquitous in indoor environments because of their use in consumer products (Wang et al., 2017). They are also found in Class B firefighting foams stored in fire departments for use on chemical fires in some states. The shift of IVOCs from the particle to the gas phase with dilution can have a large influence on their chemistry and persistence in the atmosphere. For example, in the particle phase, polychlorinated dibenzo-p-dioxins (PCDDs) persist for 1–2 weeks in the atmosphere, whereas in the gas phase, some PCDDs have atmospheric lifetimes as short as 0.5 to 2.0 days against oxidation (Table 4-1). Note that accounting for phase changes with near-field dilution in atmospheric models requires that care is taken to document dilution and temperature conditions during emissions measurements.
TABLE 4-1 Selected Major Primary Species with Toxic Potential Emitted from WUI Fires
| Transport and Partitioning | Transformation and Degradation | Atmospheric Lifetime with Respect to OH and O3 | |
|---|---|---|---|
|
Carbon monoxide (CO) |
Partitions to the atmosphere and is distributed globally via wind. CO in the troposphere is slowly transported to the mesosphere and stratosphere. |
Stable under environmental conditions. Reactions with molecular oxygen or water vapor are very slow at ambient temperatures and pressure. Primary degradation pathway of tropospheric CO is via its reaction with photochemically produced hydroxyl radicals, resulting in formation of CO2.a In the stratosphere, it reacts with atomic oxygen generated by the photodissociation of O2 to form CO2.a |
CO remains in the atmosphere for an average of about 2 months.b |
|
Formaldehyde (HCHO) |
Released to the atmosphere in large amounts and is also formed in the atmosphere by the oxidation of hydrocarbons. Efficiently transferred into clouds, rain, and surface water. Dry deposition and wet removal half-lives are estimated to be 19 and 50 hours, respectively.a |
Breaks down in the gas phase to formic acid and CO. Reacts via direct photolysis and hydroxyl radical oxidation. Oxidation can also occur in cloud droplets—producing formic acid (a component of acid rain).a |
The HCHO half-life due to reaction with the hydroxyl radical is approximately 16 hours in clean air and much less in polluted air and wildland fire plumes with elevated reactive oxidant concentrations.b |
|
Benzene (C6H6) |
Gas-phase aromatic compound released into the atmosphere in large amounts from wildfires and from fossil fuels.c |
Reacts with ozone and hydroxyl radicals.a |
Benzene’s half-life due to reaction with the hydroxyl radical is approximately 9 days.b |
|
Toluene (C7H8) |
Gas-phase aromatic compound released into the atmosphere in large amounts from wildfires and from fossil fuels.c |
Reacts with ozone and hydroxyl radicals.a |
Toluene’s half-life due to reaction with the hydroxyl radical is approximately 2 days.b |
|
Acrolein (C3H4O) |
Limited atmospheric transport. No partitioning from vapor phase to atmospheric particles. Removed from atmosphere by wet deposition.a |
Reacts with hydroxyl radicals. Products include CO, HCHO, glyoxal, and glycolaldehyde. In the presence of nitrogen oxides, products include peroxynitrate, NO, glycolaldehyde, malonaldehyde, and 3-hydroxypropanaldehyde. Direct photolysis is minor under ambient conditions.a |
Acrolein’s half-life due to reaction with the hydroxyl radical is approximately 5 hours in clean air and about half that time in polluted air.b |
|
Hydrogen cyanide (HCN) |
Slow degradation in air; thus, it can be transported over long distances. Largely remains in lower altitudes (troposphere); only 2% of tropospheric hydrogen cyanide is transferred to the stratosphere. Water-soluble cyanide particles are expected to be removed by both wet and dry deposition.a |
Resistant to photolysis. Reacts with hydroxyl radicals to form CO and NO. This photooxidation occurs at least an order of magnitude faster at lower altitudes (0–8 km) than at upper tropospheric altitudes (10–12 km).a |
The residence time of HCN in the atmosphere has been estimated to be approximately 1.3–5.0 years, depending on the hydroxyl radical concentration. |
| Transport and Partitioning | Transformation and Degradation | Atmospheric Lifetime with Respect to OH and O3 | |
|---|---|---|---|
|
Ammonia (NH3) |
The pH and temperature influence transport and partitioning. Dry deposition of NH3 dominates when there are large NH3 emissions. Where NH3 emissions are lower, wet deposition of neutralized ammonium aerosols dominates.a |
Neutralizes acidic air pollutants (e.g., H2SO4, HNO3, or HCl) to form ammonium salts in aerosols. |
The half-life for ammonia in the atmosphere is estimated to be a few days; however, it will dynamically partition very quickly between the gas phase (as ammonia) and the particle phase (as ammonium), depending on the acidity of the particles in the atmosphere. |
|
Nitrogen oxides (NOx) |
Dominant source of NOx in the air is combustion; 90–95% of NOx molecules are emitted as NO and 5–10% as NO2; however, NO is rapidly oxidized to NO2. Wet precipitation and dry deposition remove NOx from the atmosphere (US Environmental Protection Agency, 2021). |
NOx contributes to the formation of photochemical smog and acid rain (US Environmental Protection Agency, 2021). Oxidation products of NOx in the troposphere include HNO3, HO2NO2, HNO2, peroxyacylnitrates, N2O5, nitrate radical (NO3), and organic nitrates. Dominant form of NOx (NO, NO2, HNO3, etc.) in the lower atmosphere varies, depending on sunlight intensity, temperature, pollutant emissions, pollutant emission lag time, and meteorological factors. |
This group of compounds is characterized by relatively short atmospheric lifetimes of hours to days; NO and NO2 react very quickly with ozone, reaching a dynamic equilibrium. |
|
Sulfur dioxide (SO2) |
Oxidized rapidly in atmospheric waters and removed from the atmosphere by precipitation or dry deposition, mainly as sulfuric acid (acid rain). |
Oxidized by OH radicals in the gas phase and in clouds/fogs by peroxides, NO2, and transition metals, or catalytically on surfaces.a Reaction forms SO3 and subsequently sulfate.a |
Depending on the hydroxyl radical concentration, sulfur dioxide may rapidly be converted to sulfuric acid, and partitions into the particle phase. This process has a half-life of hours to days at typical hydroxyl radical concentrations. When conditions are favorable, aqueous oxidation can be considerably faster. |
|
Hydrogen sulfide (H2S) |
Partitions to surface water, groundwater, or moist soil via sorption.a |
Oxidized by oxygen to give sulfur dioxide (SO2) and ultimately sulfate compounds. Not expected to undergo significant photolysis.a |
H2S has lifetimes in air ranging from approximately 1 day in the summer to 42 days in the winter. |
|
Hydrogen chloride (HCl) |
Acts as a reservoir species, temporarily removing chlorine radicals from a catalytic ozone destruction cycle. Can be found in the gas phase, clouds, and aerosols, dependent on temperature, humidity, and ammonia level. Lower ambient temperature and higher relative humidity favor aerosol formation.a |
Can react with hydroxyl radicals to form chloride radicals and water. Chloride radicals can deplete O3 and react rapidly with VOCs. Oxidation of VOCs by chloride radicals can lead to secondary aerosol production.a |
The half-life for HCl in the atmosphere is estimated to be a few days; however, it will dynamically partition very quickly between the gas phase and the particle phase, depending on the concentration of ammonia and available cations such as sodium. |
|
Hydrogen bromide (HBr) |
Limited data on HBr in the atmosphere. Likely to be removed by both wet and dry deposition. Both acidic bromine and particulate bromine are observed in the troposphere.a |
Does not undergo photolysis in the troposphere. Can react with hydroxyl radicals to form bromide radicals and water. Like chlorine radicals, bromide radicals (Br·) can deplete O3 and react rapidly with VOCs. Experimental data show HBr may be indirectly formed in the upper atmosphere from organic bromine compounds (such as CH3Br) via the reaction of BrO with hydroxyl, OH, which then contributes to the depletion of ozone.a |
The half-life for HBr in the atmosphere is estimated to be a few hours; however, it will dynamically partition very quickly between the gas phase and the particle phase, depending on the concentration of ammonia and available cations such as sodium. |
|
Hydrogen fluoride (HF) |
Absorbed by atmospheric water (rain, clouds, fog, snow, aerosol liquid water), forming aqueous hydrofluoric acid. Removed by dry and wet deposition (i.e., acid precipitation). Can be neutralized (e.g., by sodium or other cation) and taken up by particulate matter or dust.a |
HF is much too reactive to reach the upper atmosphere, and tropospheric sources therefore do not interfere with the ozone layer. Polymerization or depolymerization reaction of HF does not destroy or remove HF or its oligomers (dimer (HF)2, hexamer (HF)6, and octamer (HF)8) from the air.a |
The atmospheric lifetime of HF is less than 4 days. Deposition is the dominant mechanism driving removal from the atmosphere (Cheng, 2018). |
|
Per- and polyfluoroalkyl substances (PFASs, e.g., perfluorooctane sulfonate, perfluorooctanoic acid) |
Found in both the gas and particle phases. Removed via wet and dry deposition.a |
PFASs include thousands of compounds across several compound classes. Perfluorinated carboxylic acids and sulfonates are resistant to photolysis and atmospheric photooxidation, but fluorotelomer and fluorosulfamido alcohols can be oxidized by OH radicals to form carboxylic acids and sulfonates (Ellis et al., 2004; Young et al., 2007) |
The lifetimes are days to weeks (Ellis et al., 2003). |
| Transport and Partitioning | Transformation and Degradation | Atmospheric Lifetime with Respect to OH and O3 | |
|---|---|---|---|
|
Polycyclic aromatic hydrocarbons (PAHs) |
Subject to short- and long-range transport and are removed by wet and dry deposition onto soil, water, and vegetation. Partitioning of PAHs between the gas and the condensed phases depends on their volatility, the temperature, and the concentration of particles in the atmosphere. PAHs having two to three rings are present in air predominantly in the vapor phase; those with four rings exist both in the vapor and particulate phase; and those having five or more rings are found predominantly in the particle phase. Atmospheric residence time and transport distance depend on the size of the particles to which the PAHs are sorbed and on climatic conditions (which will determine rates of wet and dry deposition). Particles with a diameter range of 0.1–3.0 μm, with which airborne PAHs are principally associated, remain airborne for 5–10 days and can be transported long distances. |
Two types of chemical reactions transform PAHs: (1) reactions between PAHs adsorbed on particle surfaces and oxidant gases like NO2, O3, and SO3, and (2) gas-phase photooxidation producing oxidized derivatives such as quinones, ketones, or acids. Photochemical reactions involve NOx, N2O5, OH, O3, SO2, and peroxyacetyl nitrate. Reactions of PAHs, including fluoranthene and pyrene, with the OH radical (in the presence of NOx) and with N2O5 lead to the formation of nitroarenes in the ambient air, which are more mutagenic than their parent PAHs. Reaction with ozone or peroxyacetylnitrate yields diones; nitrogen oxide reactions yield nitro- and dinitro-PAHs. Compounds adsorbed to particles are more resistant to photochemical reactions than gas-phase PAHs.a Variations in chemical composition of different types of particles such as diesel exhaust and wood smoke might strongly affect the reactivity of PAHs.a |
The atmospheric lifetimes of naphthalene and phenanthrene, due to reaction with the hydroxyl radical in relatively clean air, are about 6 hours and 10 hours, respectively; PAHs may be more resistant to oxidation if sorbed onto particles.b |
|
Polychlorinated biphenyls (PCBs) |
Enter the atmosphere when volatilized from soil and water. In the atmosphere, PCBs are present both in the vapor phase and sorbed to aerosol particles. Transport and partitioning behavior of PCBs depends on the number of chlorines on the biphenyl molecule, because of the influence on compound vapor pressure. Biphenyls with 0–1 chlorine atoms remain in the atmosphere, while those with 1–4 chlorines gradually migrate toward polar latitudes in a series of volatilization/deposition cycles, those with 4–8 chlorines remain in the mid-latitudes, and those with 8–9 chlorines remain close to the source of contamination. PCBs in the vapor phase are more mobile and transported farther than particle-bound PCBs. |
The ability of PCBs to be degraded or transformed in the environment depends on the degree of chlorination of the biphenyl molecule as well as on the isomeric substitution pattern. The vapor-phase reaction of PCBs with hydroxyl radicals is the dominant transformation process in the atmosphere. Possible reaction schemes involve the formation of a 2-hydroxybiphenyl intermediate, which quickly degrades to chlorinated benzoic acid. Insufficient data are available on the importance of photolysis and/or chemical reactions of particle-phase congeners.a |
The calculated tropospheric lifetime for the reaction of PCBs increases as the number of chlorine substitutions increases. The tropospheric half-lives are 2 days for monochlorobiphenyls, 4 days for dichlorobiphenyls, 10 days for trichlorobiphenyls, and 15 days for tetrachlorobiphenyls; PCBs may be more resistant to oxidation if sorbed onto particles.b |
|
Heavier and coplanar PCBs tend to be particle bound and/or more readily degraded in the atmosphere. PCBs are removed by wet deposition; by dry deposition of aerosols; and by vapor adsorption or partitioning to water, soil, and plant interfaces.a |
|||
|
Polychlorinated dibenzo-p-dioxins (PCDDs) |
Combustion-generated PCDDs associated with particulate matter can be distributed over large areas. During transport, PCDDs partition between the vapor phase and particle-bound phase. However, partitioning is largely to the particulate phase. Removed from the atmosphere via wet deposition, particle dry deposition, and gas-phase dry deposition. The less chlorinated PCDD congeners (tetra-CDD and penta-CDD) occur in greater proportion in the vapor and dissolved phases of air and rain, whereas the more chlorinated congeners (hepta-CDD and octa-CDD) are associated with particles.a |
PCDDs’ dominant transformation processes are photolysis and gas-phase diffusion, and volatilization with subsequent photolysis. Transformation reactions depend on whether the PCDD is in the vapor or particulate phase. Vapor-phase PCDDs are not likely to undergo reactions with atmospheric ozone, nitrate, or hydroperoxy radicals; however, reactions with hydroxyl radicals may be significant, particularly for the less-chlorinated congeners (up to tetra-CDD). Based on the photolysis lifetimes of PCDDs in solution, it is expected that vapor-phase PCDDs will also undergo photolysis in the atmosphere, although reactions with hydroxyl radicals will predominate. In the air, the low vapor pressure of octa-CDD results in its partitioning primarily to the particulate phase rather than the vapor phase; therefore, atmospheric photodegradation is less likely to occur for this tightly bound congener.a |
The atmospheric lifetimes of PCDDs are estimated to range from 0.5 days for mono-CDD to 9.6 days for octa-CDD, with tetra-CDD having a lifetime of 0.8–2 days. Particulate-bound PCDDs are removed by wet or dry deposition with an atmospheric lifetime of approximately 10 days and, to a lesser extent, by photolysis. |
|
Polybrominated diphenyl ethers (PDBEs) |
Exist in both gas and particle phases in the atmosphere. Particulate-phase PBDEs are removed from the atmosphere by wet and dry deposition.a |
Vapor-phase PBDEs may be degraded via oxidation with hydroxyl radicals or direct photolysis, with photolysis being the dominant method. Limited data are available on the rate and extent of photolysis of PBDEs in air.a |
The half-lives for this reaction in air are estimated to be 29, 140, and 476 days, respectively, for penta-, octa-, and deca-BDE homologs, calculated using a structure estimation method. |
|
Phosphate ester flame retardants |
Can be present in the gas or particle phase. Subject to wet and dry deposition.a |
May be degraded through reaction with hydroxyl radicals. Semi-volatile phosphate esters have the potential to hydrolyze to diesters, monoesters, and phosphoric acid.a |
Atmospheric half-lives are on the order of 1–12 hours for the selected phosphate esters. Particle-bound phosphate esters are more resistant to OH oxidation with lifetimes of 4–14 days (Liu et al., 2014). |
NOTES: Based on laboratory and field measurements from wildland fires, WUI fires, and structure fires, as well as information about materials. HCl, HBr, HF, PFASs, PCBs, PCDDs, PDBEs, and phosphate ester flame retardants are expected to be found in higher concentrations in WUI fires than in wildland fires.
a US Agency for Toxic Substances and Disease Registry. 2021. https://www.atsdr.cdc.gov/ (accessed February 26, 2022).
b US Environmental Protection Agency. 2021. EPI Suite™-Estimation Program Interface. https://www.epa.gov/tsca-screening-tools/epi-suitetm-estimation-program-interface (accessed December 22, 2021).
Recent progress has been made in quantifying IVOCs in particulate emissions from wildland fires and predicting their downwind chemistry in models (Hatch et al., 2017; Theodoritsi et al., 2021; Yee et al., 2013). However, the characterization of IVOC emissions from burning structures in WUI fires is limited, and predictive modeling has not yet explored the implications of the near-field phase changes and subsequent local- to regional-scale chemical processing of these species. Measurements provide limited evidence for the enhancement of structure-related tracer species above the ambient background in regional measurements; however, some of these species or their reaction products may persist over regional scales (Table 4-1).
Formation of Secondary Species with Toxic Potential
While concentrations of primary species with toxic potential decrease with distance downwind as a result of dilution with ambient air, wet and dry deposition, and oxidation (Table 4-1), oxidation can also produce secondary species with toxic potential (Table 4-2). Measurement of tracer compounds that are specific to WUI fire emissions (primary tracers) and WUI chemistry (secondary tracers) could be used to identify and understand the influence of WUI fires on downwind communities. While little is known about secondary species with toxic potential from WUI fires, elevated concentrations of the hydroxyl radical, reactive nitrogen, reactive chlorine, and transition metals are likely to exist in WUI plumes (see “Major Atmospheric Oxidants Driving Gas-Phase Chemistry” below) and will react with a variety of unsaturated organic compounds to form more oxygenated, chlorinated, and nitrated products that may have increased toxicity (Tuet et al., 2017; Wong et al., 2019). For example,
- Nitrophenols are likely to form in WUI fire plumes since phenols are components of wildland fire plumes and nitrophenols are known to form through daytime and nighttime gas-phase radical chemistry and through reactive nitrogen (e.g., N2O5, ClNO2) chemistry in aqueous or multiphase reactions in the atmosphere (Harrison et al., 2005).
- Chlorinated and oxygenated aromatics are likely to form in WUI fire plumes, since aromatic compounds are substantial components of wildland fire plumes, chlorine radicals and hydroxyl radicals are expected to be present in WUI fire plumes, and chlorinated aromatics and oxygenated aromatics are known to form in the atmosphere (e.g., from gas-phase chlorine radical reaction with PAHs; Ohura et al., 2005; Riva et al., 2015).
TABLE 4-2 Examples of Secondary Species with Toxic Potential Observed or Proposed to Occur in WUI Fires
| Compound or Compound Class | Precursor | Formation Pathway |
|---|---|---|
| Ozone | NOx and VOCs | Gas-phase oxidation of VOCs in the presence of NOx and ultraviolet light |
| Secondary PM2.5 | SO2, H2S, NOx, NH3, VOCs, and IVOCs | (1) Gas-phase oxidation followed by vapor pressure–driven partitioning into the particle phase; (2) aqueous or multiphase reaction in aerosol, clouds, or fogs followed by water evaporation and retention of lower volatility products in the particle phase; (3) heterogeneous or catalytic surface reactions on aerosol particles |
| Formaldehyde, other aldehydes | Most VOCs | Gas phase oxidation of VOCs (Luecken et al., 2018) |
| Nitroaromatics | Aromatics | Gas-phase and aqueous or multiphase chemistry with reactive nitrogen species (e.g., nitrophenols; Harrison et al., 2005; Pang et al., 2019) |
| Chlorinated aromatics | Aromatics | From chlorine radical reaction (e.g., chlorinated PAHs; Ohura et al., 2005; Riva et al., 2015) |
| Perfluorocarboxylic acids | Fluorotelomer alcohols | OH oxidation in gas phase (e.g., perfluorooctanoic acid from 8:2 fluorotelomer alcohol; Wallington et al., 2006) |
NOTE: Based on observation in wildland fires or WUI fires, or knowledge about WUI materials or emissions and fundamental atmospheric chemistry knowledge.
- To the extent that PFASs are emitted from WUI fires, oxidation may yield perfluorocarboxylic acids. Photochemical hydroxyl radical oxidation of fluorotelomer alcohols (a type of PFAS found in homes; Hall et al., 2020; Sha et al., 2018), where present, yields perfluorocarboxylic acids such as perfluorooctanoic acid (Wallington et al., 2006).
Stable unique products of these chemistries (e.g., products of chlorine radical chemistry; tracers of synthetic polymer degradation products) could be used as chemical tracers to identify the influence of secondary formation, understand how human-made materials alter the chemistry, and determine the dominant daytime and nighttime oxidants, and potentially to identify dominant formation pathways (e.g., gas-phase chemistry vs aqueous or multiphase chemistry).
Wildland fire emissions are known to increase formation of ozone, a secondary species with toxic potential formed through photochemical reactions involving oxides of nitrogen and VOCs (Xu et al., 2021); insights pertaining to ozone formation in WUI fires are provided in the section called “Ozone Formation,” below. Likewise, secondary PM2.5 will also form downwind of WUI fires. Particulate sulfate forms from the oxidation of SO2 and H2S in the gas phase, in clouds and fogs, or in some cases on catalytic surfaces to form sulfuric acid. Sulfuric acid or its neutralized salt (e.g., ammonium sulfate) has a low vapor pressure and thus condenses on existing particles or forms new particles (Seinfeld and Pandis, 2016). Particulate nitrate forms from the oxidation of oxides of nitrogen to nitric acid and the subsequent neutralization of nitric acid by ammonia (Seinfeld and Pandis, 2016). As with sulfate, secondary organic aerosol can form through gas-phase and aqueous reactions; its formation is described in detail in the “Secondary Organic Aerosol Formation” section below. As the aerosol ages, primary and secondary PM2.5 components are frequently found in the same particles (i.e., internally mixed). However, the components are often phase separated, with sulfate, nitrate, and associated water in the core and organics in the shell, except at high relative humidities when the water content of the aerosol particles is substantial and liquid-like components can become homogeneously mixed (Ciobanu et al., 2009; Li et al., 2021; Zuend and Seinfeld, 2012).
Much is left to learn about the formation of secondary species with toxic potential downwind of WUI fires. An unresolved question is whether the “per unit mass” toxicity of the WUI fire plume mixture increases or decreases with distance downwind. O’Dell et al. (2020) found that hazard ratios and cancer risks associated with gas-phase hazardous air pollutants derived from wildland fires decreased with distance downwind during the Western Wildfire Experiment for Cloud Chemistry, Aerosol Absorption, and Nitrogen (WE-CAN) campaign. The largest hazardous air pollutant contributors were acrolein, hydrogen cyanide, and formaldehyde. Other investigators observed an increase in the oxidative potential (per unit mass) of organic aerosol when smoke from wildland fire fuels was oxidized (aged), suggesting that the potential of organic aerosol to play a role in oxidative stress may also increase with atmospheric aging (Wong et al., 2019). Little understanding exists regarding how far from the immediate fire zone human-made materials alter the toxicity of the air pollution mixture, and a more comprehensive assessment of the evolution of primary and secondary WUI fire plume toxicants is needed to understand the evolution of health impacts with atmospheric aging.
Finding: The dominant health-relevant secondary species (secondary species with toxic potential) associated with WUI fires have not been definitively identified, and more work is needed to characterize species with toxic potential.
Finding: It is not well understood how and how far downwind human-made materials influence the composition of WUI fire plumes.
Research need: Research is needed to identify compounds that can serve as tracers of (1) a WUI fire’s influence on an air mass (e.g., aerosol tracers), (2) the influence of human-made materials on the chemistry, (3) the influence of particular oxidants, and (4) the influence of specific types of chemistry (e.g., multiphase chemistry) in WUI fire plumes.
Major Atmospheric Oxidants Driving Gas-Phase Chemistry
OH radicals, ozone, nitrate radicals, and chlorine radicals are expected to be the main atmospheric gas-phase oxidants in WUI fire plumes, although other halogens may also play a role. Much of this expectation comes from the current knowledge of WUI fire emissions, fundamental atmospheric chemistry, wildland fire studies (e.g., the recent WE-CAN and Fire Influence on Regional to Global Environments and Air Quality [FIREX-AQ] studies), and studies of atmospheric halogen chemistry in industrial areas. Coordinated measurements, including the use of tracers of chemical processes, have played an important role in improving our understanding of atmospheric chemistry. Atmospheric oxidants transform the smoke mixture as it travels downwind, forming more oxidized and functionalized gases and secondary organic aerosols (e.g., wildland fire aerosol; Zhou et al., 2017).
OH radicals (daytime), nitrate radicals (nighttime), and ozone are mainly responsible for oxidation of organic gases in wildland fire plumes; the implications for oxidation by compound class are provided for wildfires in the FIREX-AQ study by Decker et al. (2021). HONO and HCHO photolysis provides a rapid daytime source of OH radicals (Theys et al., 2020; Veres et al., 2010). In fact, Theys et al. (2020) argue, based on satellite-derived concentrations, that globally, HONO emissions are responsible for two-thirds of OH production in fresh wildland fire plumes, although their importance drops off rapidly as the plume ages (Xu et al., 2021). OH radical concentrations that are 5–20 times higher than those in background air have been measured in wildland fire smoke (Hobbs et al., 2003; Yokelson et al., 2009). Nitrate radical, produced from the oxidation of NO2 by O3, is a major nighttime oxidant in wildland fire plumes and may be enhanced in WUI fires due to high emissions of oxides of nitrogen formed as a result of nitrogen in the fire’s fuel (fuel NOx). However, the NO3 radical is rapidly lost to photolysis during the day (Ng et al., 2017). Oxygenated aromatics in wildland fires are highly reactive with the NO3 radical (Akherati et al., 2020; Decker et al., 2019). A wildland fire oxidant reactivity budget quantifying the roles of OH (daytime), NO3 (nighttime), and O3 (both) through the use of aircraft measurements has been provided by Decker et al. (2021).
The role of halogens in WUI fire plume chemistry warrants additional consideration because of the increase in both halogen and fuel NOx emissions in these fires (Chapter 3). HCl and particulate chlorine are emitted from wildland fires (McMeeking et al., 2009), but HCl production is increased by the decomposition of polymers from the burning of structures (Chapter 3). Cl radicals are formed at the near-field scale through mechanisms described in Chapter 3. Under atmospheric conditions that lead to cycling between reactive and nonreactive forms of nitrogen (e.g., conditions with high concentrations of N2O5), the impact of Cl radicals on chemistry persists with regional transport. Multiphase or aqueous reactions with N2O5 release chlorine (as ClNO2) from aerosols during transport, and Cl radicals are subsequently formed via daytime photolysis (Ahern et al., 2018; Goldberger et al., 2019; Thornton et al., 2010).
The Cl radical is roughly an order of magnitude more reactive with many VOCs than the OH radical (Faxon and Allen, 2013). It fragments, oxidizes, and sometimes forms chlorinated organics, and it produces OH radicals (Faxon and Allen, 2013). The OH-to-Cl radical ratio is high enough that OH is the dominant oxidant in most cities (e.g., 9 percent of the primary radical source in Los Angeles; Young et al., 2014). However, the chlorine radical can be an important oxidant in several continental locations (e.g., the Houston, Texas, ship channel; Riemer et al., 2008; Tanaka et al., 2003). Young et al. (2014) conclude based on chemical modeling that the direct influence of the Cl radical on VOC oxidation is evident (e.g., in VOC tracer ratios) when OH-to-Cl radical ratios are less than 200, but that the true impact of the Cl radical is greater because of secondary reactions and the recycling of reactive chlorine.
It is logical that chlorine and potentially other halogens would play an enhanced role in WUI fire plumes. A chlorine radical reactivity budget for WUI fires is needed to better assess the impact of this potentially important radical species. In addition to HCl, HBr is also emitted from structure fires (Chapter 3), yet the contribution of reactive bromine chemistry is not understood.
Finding: Coordinated measurements, including the use of tracers of chemical processes, have proven useful in improving our understanding of the chemical evolution in aging wildland fire plumes but are much more limited for WUI fires.
Finding: Current understanding of the dominant daytime and nighttime oxidant species, including an assessment of any enhanced role of halogen chemistry, is incomplete; this information is important to estimate the atmospheric lifetime of primary species with toxic potential and predict the formation of secondary species with toxic potential downwind of WUI fires.
Research need: Field measurements and controlled experiments are needed to identify the dominant daytime and nighttime atmospheric oxidants, their concentration ranges, and the influence of emissions from human-made materials on oxidation; this information is important to determine typical atmospheric lifetimes for primary species with toxic potential and better predict concentrations of secondary species with toxic potential in WUI fire plumes.
Ozone Formation
Wildland fires have a substantial impact on regional summertime ozone concentrations (and exposures) in the western United States due to their substantial emissions of ozone precursors (i.e., VOCs and NOx; Friedli et al., 2001; Koss et al., 2018; Xu et al., 2021). Lu et al. (2016) attribute one-third of summer days exceeding the National Ambient Air Quality Standard for ozone in the western United States to wildland fires (daily maximum 8-hour average, MDA8, ozone concentrations exceeding 70 ppbv). Wildland fire MDA8 ozone increases can be as high as 5–40 ppbv on some days (Dreessen et al., 2016; Gong et al., 2017; Lu et al., 2016), and the majority of observations suggest at least some ozone increase (Jaffe and Wigder, 2012). VOC-to-NOx ratios are approximately 10–30 for temperate wildland fires, suggesting that NOx is typically the limiting precursor after the first few hours of rapid formation and transport (Akagi et al., 2011; Andreae, 2019; Jaffe and Wigder, 2012; Jaffe et al., 2020; Xu et al., 2021). Thus, the partitioning of reactive nitrogen between NOx, NO3, peroxyacetyl nitrate, and other N species will regulate ozone production, and ozone production may increase where the wildland fire plumes interact with NOx-rich pollution (e.g., when they encounter cities and powerplant plumes). Peroxyacetyl nitrate acts as a long-range NOx reservoir species, enabling ozone formation to continue over hundreds of kilometers (Alvarado et al., 2010; Briggs et al., 2017).
It is possible that increased reactive nitrogen and chlorine emissions in WUI fires may drive faster ozone formation (e.g., by increasing OH) and more ozone production (e.g., via NOx and Cl reservoir species) at a regional scale. However, the magnitude of these effects is not known. Ozone formation is a photochemical process, and thus it also depends on the photolysis rate, which can be reduced by aerosol extinction (e.g., the rate can be reduced by 10–20 percent; Real et al., 2007). Likewise, clouds and wet aerosols can reduce ozone formation by removing water-soluble ozone precursors (Yokelson et al., 2003).
Finding: The increase in reactive nitrogen and halogen emissions in WUI fires relative to wildland fires, and the VOC-to-NOx ratios of WUI fire emissions, are not well characterized; this information could improve prediction of ozone from WUI fires.
Secondary Organic Aerosol Formation
Aircraft, remote sensing, field, and laboratory studies provide insights into secondary organic aerosol (SOA) formation from wildland fire emissions (Hodshire et al., 2019). For example,
- Garofalo et al. (2019) did not see a change in the total organic aerosol mass concentration (relative to CO) with distance downwind but did see an increase in the degree of oxygenation, as indicated by an increase in oxygenation markers and a concurrent decrease in the biomass burning marker.
- Palm et al. (2020) simulated the wildfire chemistry, constrained by aircraft measurements, and concluded that 87 percent of the organic aerosol formed in the atmosphere was composed of evaporated IVOCs. Phenolic SOA precursors accounted for the majority of the remaining SOA. At 3–6 hours of daytime oxidation, two-thirds of the aerosol mass was primary, and one-third was secondary.
- Through remote sensing, validated in part by aircraft measurements, Junghenn Noyes et al. (2020) documented an increasing ratio of brown carbon to black carbon aerosol with distance downwind of the Government Flats Complex Fire in Oregon, suggesting the atmospheric formation of light-absorbing organic aerosol in the plume.
SOA Formation via Vapor Pressure–Driven Partitioning
Based on fundamental principles of atmospheric chemistry, the committee expects that gas-phase oxidation via OH and Cl (daytime), NO3 (nighttime), and O3 results in fragmentation of some WUI fire organics and functionalization of others. Thus, gas-phase oxidation can lead to the formation of smaller, more oxygenated (water-soluble) gas-phase compounds. However, when oxidation leads to functionalization and the products have a low enough vapor pressure, they condense (sorb into preexisting aerosol) and form a SOA (Kroll and Seinfeld, 2008). SOA formation through vapor pressure–driven partitioning happens on a timescale of minutes to hours (Palm et al., 2020). SOA formed from wildland fires via vapor pressure–driven partitioning typically has a lower volatility and higher O-to-C ratios than primary organic aerosol emitted by wildland fires (Garofalo et al., 2019; Palm et al., 2020).
Oxygenated aromatic compounds and IVOCs are understood to be the main SOA precursors through gas-phase oxidation and vapor pressure–driven partitioning (Akherati et al., 2020; Chan et al., 2009; Coggon et al., 2019; Patoulias et al., 2021; Posner et al., 2019; Tkacik et al., 2012; Yee et al., 2013; Zhao et al., 2014b). These are among the SOA precursors in WUI fire plumes. However, model prediction of the magnitude of SOA formation downwind of wildland fires (Theodoritsi et al., 2021) and WUI fires remains uncertain due in part to inadequate chemical characterization of IVOCs and other precursor emissions, the partial understanding of oxidation mechanisms, and a lack of thermodynamic data for the oxidation products. Phenol, guaiacol, dioxins, and synthetic polymer degradation products are among the potential WUI precursors to SOA formation through gas-phase oxidation and vapor pressure–based partitioning.
SOA Formation via Aqueous, Heterogeneous, or Multiphase Chemistry
Chemistry in WUI fire plumes can also occur on the surfaces of aerosol particles (heterogeneous) and in the liquid water contained in clouds, fogs, and aerosol particles (multiphase or aqueous). Atmospheric liquid water and water-soluble compounds are abundant in wildland fire plumes. Formaldehyde, isocyanic acid, glycolaldehyde, acetic acid, phenols, furfurals, and amines, which are found in wildland fire plumes (Friedli et al., 2001; Hatch et al., 2015; Koss et al., 2018; Leslie et al., 2019; McFall et al., 2020; Permar et al., 2021; Schauer et al., 2001; Selimovic et al., 2018; Tomaz et al., 2018), for example, will partition into clouds and wet aerosols, where they can react further through radical or non-radical reactions (Bianco et al., 2020). In some cases, products of aqueous organic chemistry remain in the particle phase after water evaporation (e.g., Brégonzio-Rozier et al., 2016; Lee et al., 2011), and water evaporation itself can drive accretion reactions that enhance particle-phase retention (e.g., for aldehydes and amines; Hawkins et al., 2014; Loeffler et al., 2006). Thus, chemistry in clouds and wet aerosols can lead to “aqueous” SOA formation (Blando and Turpin, 2000; Lamkaddam et al., 2021; McNeill, 2015; Tilgner et al., 2013; Volkamer et al., 2009).
Because the precursors are small and oxidized, with high O-to-C ratios, the SOA produced also has high O-to-C ratios. In clouds, radical reactions dominate, whereas in wet aerosols, non-radical reactions can be important (Lim et al., 2010; McNeill, 2015). For example, peroxides and epoxides from isoprene oxidation undergo rapid reactive uptake via acid catalysis in wet aerosols (Surratt et al., 2010); this multiphase isoprene chemistry is a major source of SOA in the southeastern United States (Budisulistiorini et al., 2013; Ying et al., 2015). The aqueous oxidation of particulate organics in clouds and wet aerosols can also result in volatile losses of oxidation products—a reduction in organic aerosol mass.
Several studies provide evidence for the influence of atmospheric aqueous chemistry on wildland fire smoke evolution:
- Decreasing concentrations of methanol and increasing concentrations of formaldehyde above clouds affected by savanna fires have been attributed to in-cloud oxidation of savanna fire emissions (Tabazadeh et al., 2004; Yokelson et al., 2003).
- Zhang et al. (2017) measured an increase in the aerosol brown-carbon-to-black-carbon ratio in the outflow, compared to inflow, of convective clouds impacted by a wildland fire plume, suggesting aqueous SOA formation.
- Gilardoni et al. (2016) observed the formation of a light-absorbing SOA in fog and wet aerosol impacted by residential woodburning and estimated that aqueous chemistry accounts for approximately one-half of the SOA mass in the Po Valley, Italy, in a typical winter (Gilardoni et al., 2016; Paglione et al., 2020).
- Lin et al. (2010) concluded that cloud processing of straw-burning emissions could explain high correlations between cloud chemistry tracers (i.e., oxalate and sulfate) and humic-like substances in ambient aerosols affected by the field burning of rice straw.
- Cook et al. (2017) observed light-absorbing aqueous-phase products of syringol and guaiacol oxidation in wildland fire–influenced cloud water samples.
- Tomaz et al. (2018) observed the formation of known SOA components upon aqueous OH radical oxidation of emissions from burning mixtures of forest materials.
- The aqueous SOA formation potential of several wildland fire emissions (e.g., acetic, glycolic, pyruvic, succinic, glutaric, adipic, and lactic acids; glycolaldehyde; phenol; catechol; guaiacol; vanillin; levoglucosan) and the aqueous formation of nitrophenols among other products have been studied in the laboratory (Carlton et al., 2006; Chang and Thompson, 2010; Charbouillot et al., 2012; Li et al., 2014; Lim et al., 2013; Pang et al., 2019; Perri et al., 2009; Sun et al., 2010; Tan et al., 2012; Vidović et al., 2020; Yu et al., 2014, 2016; Zhao et al., 2014a).
- Models estimate in-cloud SOA from biomass burning at 20–30 Tg/yr globally (Liu et al., 2012; Lin et al., 2014).
Predictions of SOA formation through aqueous chemistry are highly uncertain (Barth et al., 2021), in part because of uncertainties in aqueous-phase oxidant concentrations. It is generally accepted that OH concentrations are depleted in clouds in comparison to equilibrium (Arakaki et al., 2013), although there is some indication that concentrations could be much higher under certain circumstances (Lamkaddam et al., 2021; Paulson et al., 2019) and that reactions at air-water interfaces can be orders of magnitude higher than in bulk water (Enami et al., 2014). Photosensitized reactions, HONO, halogens, reactive nitrogen, H2O2, and metals (Fenton chemistry) are all possible sources of aqueous-phase radicals (Bianco et al., 2020). Organics could also plausibly recycle radicals through autocatalysis (Rossignol et al., 2014). Acidity is also a critical factor in determining the partitioning of aqueous-phase products between gas and particle phases (Ortiz-Montalvo et al., 2014; Tilgner et al., 2021).
Overall, significant progress has been made in understanding the gas-phase chemistry, gas-to-particle partitioning, and SOA formation associated with the burning of biomass from fields and forests. Many of the most important processes are controlled by radical chemistry. Because WUI fires have the potential to have significantly different radical reactivities than wildland fires, due to the increased presence of halogen radicals, metals, and other species, critical chemical pathways have the potential to have different rates and to be qualitatively different than in wildland fires. More work is needed to understand WUI plume chemistry, especially the multiphase, aqueous, and catalytic chemistry. Issues include identifying the critical WUI species and processes. A combination of aircraft and other field measurements (see Chapter 7), controlled laboratory experiments, and modeling (see below) will be needed to address these questions.
Finding: Multiphase, aqueous, and catalytic chemistry in WUI fire plumes is poorly understood.
Research need: An improved understanding of key processes and formation pathways such as the roles of multiphase, aqueous, and catalytic chemistry in WUI fire plumes is needed.
CURRENT PRACTICE IN MODELING FAR PLUMES
A critical role of smoke models is to provide forecasts for public health guidance; coordinated measurements and modeling, used in conjunction with controlled experiments, also test our understanding of emissions and atmospheric processing, and enable model refinements. The physical movement of wildland fire plumes over
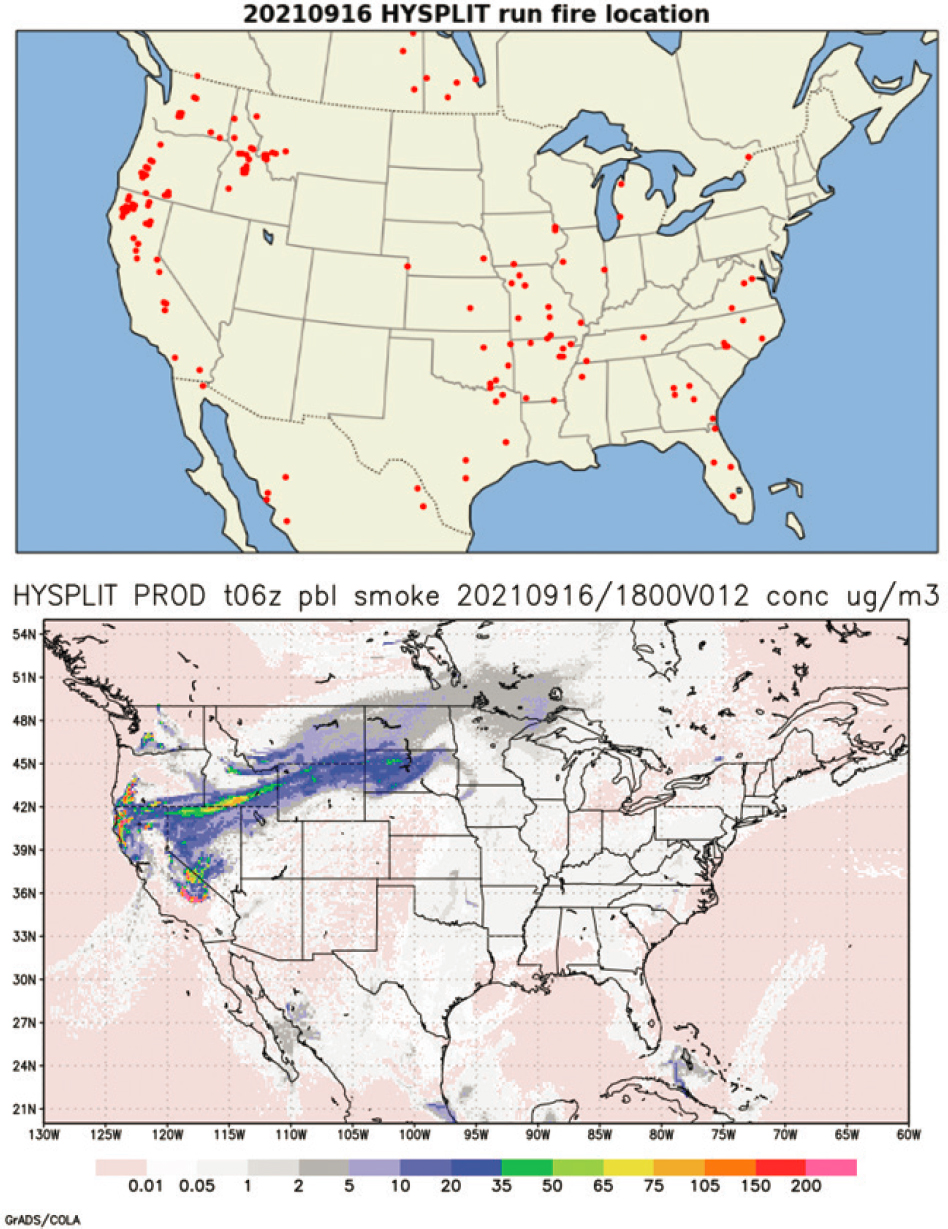
length scales of hundreds to thousands of kilometers is typically modeled using trajectory or dispersion models. These types of models typically model few or none of the chemical transformations that occur in wildland fire and WUI plumes. They do, however, predict dilution, some physical processes such as deposition, and the direction of movement of plumes. These features make transport and dispersion models useful for predicting likely atmospheric concentrations of particulate matter and are used in public notification of risk (activities to avoid). For example, the smoke forecasting system from the National Oceanic and Atmospheric Administration uses National Weather Service inputs from the North American Mesoscale model and smoke dispersion simulations from the Hybrid Single-Particle Lagrangian Integrated Trajectory model (Stein et al., 2015) to produce a daily prediction of smoke transport and concentration (NOAA ARL, 2012). As an example, Figure 4-2 shows fire location (top) and fire plume projections (bottom) for September 16, 2021, when fire plumes from Idaho and California extended east as far as Minnesota (NOAA ARL, 2021).
The accuracy of these models of plume trajectories is dependent on the ability to accurately assess the vertical height of the plume, since wind speeds and direction can vary significantly with height. Paugam et al. (2016) provide a review of plume rise performance in chemical transport models along with the atmospheric and fire parameters governing plume rise and assess the sensitivity of dispersion predictions to the predicted plume rise. More recent tools (https://hwp-viz.gsd.esrl.noaa.gov/smoke/; https://rapidrefresh.noaa.gov/hrrr/HRRRsmoke/) that make use of fire radiative power data to estimate plume rise have provided significant advances in trajectory modeling of wildfires (Ahmadov et al., 2017).
In an evaluation of the forecasting ability of 12 smoke models for a case study on a single wildland fire, Ye et al. (2021) found that all models underpredicted the amount and extent of the plume, while predictions of PM2.5 were biased in both directions, depending on the model. The authors attributed biases to uncertainties in emissions and plume rise, although atmospheric chemistry could certainly also play a role. This study demonstrates challenges involved in smoke forecasting and provides some insight into where additional effort is needed to improve prognostic modeling for WUI fires. Comprehensive summaries of smoke models can be found in the literature (Jaffe et al., 2020; Ye et al., 2021).
Finding: Models are currently available and can be useful for predicting likely impacts, public notification of risk (activities to avoid), and forecasting of particulate matter concentrations.
Research need: Continued improvement is needed in smoke dispersion models for use in both retrospective and prospective modes.
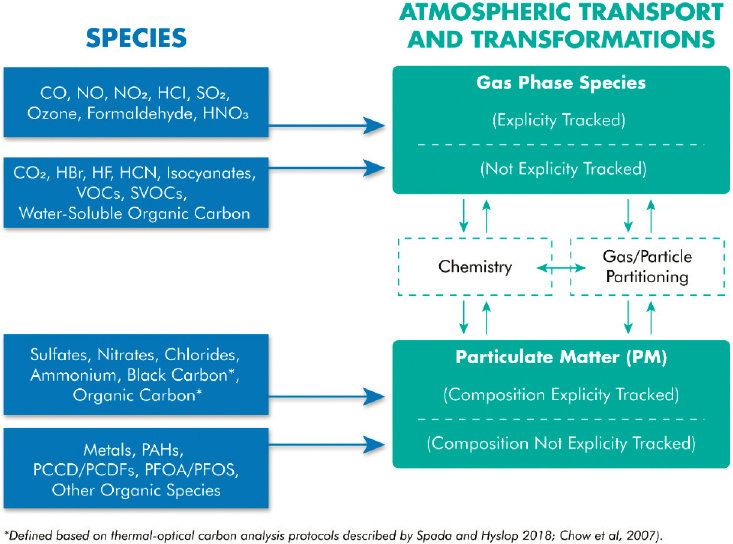
Different types of models are used to characterize chemical transformations in wildland fire and WUI plumes over local, regional, and continental scales. The complex gas-phase chemical transformations, particle-phase chemical transformations, and gas-to-particle partitioning associated with wildland fires and WUI fires, outlined in this chapter, are tracked using computationally intensive models that couple chemical transport and transformation. These modeling tools are broadly referred to as chemical transport models, and include modeling frameworks such as the Community Multiscale Air Quality (CMAQ) Modeling System (https://www.epa.gov/cmaq) and the Comprehensive Air Quality Model with Extensions (https://www.camx.com/). Because these models are so computationally intensive, simplified gas-phase chemical mechanisms, particle-phase chemical mechanisms, and models of gas-to-particle partitioning processes are used in the models. These “reduced” mechanisms track a limited number of molecular species and compound classes explicitly. The majority of primary and secondary pollutants are grouped into very broad “lumped” chemical classes such as inorganic compounds, VOCs, semi-VOCs, water-soluble organic compounds, and particulate matter. The general relationships between lumped and explicitly tracked molecular species in predictive chemical transport models, and unresolved species, is illustrated in Figure 4-3.
Chemical transport models are used to couple the modeling of transport and chemical transformation and are widely used in developing and evaluating air quality management plans for reducing ozone, particulate matter, and other pollutants in urban and regional atmospheres. Chemical transport models have also been applied to examine the coupled transport and chemical transformation of fire plumes, most commonly in retrospective analyses of wildland fire and WUI fire events, rather than in forecast mode (Goodrick et al., 2012).
Chemical transport models have generally not been optimized to address the unique chemistries and transport characteristics of wildland fire plumes. Nevertheless, a number of fire episodes that have impacted air quality over regional to continental scales in the United States have been modeled (Fann et al., 2018; Huang et al., 2021; Jiang and Yoo, 2019; Wilkins et al., 2018). These simulations, when compared to ambient observations, have tended to underestimate peak ozone and particulate matter concentrations associated with fires. In addition, the simplified chemistries used in the models for computational efficiency simulate only a limited number of chemical species, and most model applications are not formulated to track toxicants generated by fires. Limitations of current-generation chemical transport modeling tools in simulating regional- to continental-scale fire events are described in more detail below.
Limitations of Condensed Chemistries
Chemical transport models use simplified representations of chemical transformations because models of the full chemistry, even if all reactions and rates were known, would be too computationally intensive to use within the context of the models. One commonly used simplification is to represent classes of compounds by a single chemical species. For example, the reactions of all substituted aromatic compounds might be represented by the reactions of one or two “lumped” species. Another commonly used simplification is to represent collections of similar functional groups in molecules by a single lumped species. For example, the reactions of all paraffinic carbons in a mixture might be represented by a single chemical species.
These “lumped” or condensed mechanisms pose multiple problems for modeling fire emissions. First, in lumping chemical species, information about specific molecular concentrations, which is needed for estimating concentrations of toxicants, is lost. New condensed mechanisms with the ability to track key individual chemical species could be developed.
A second, and related, problem is that changing individual reactions in condensed mechanisms is problematic. As a simple example, consider a condensed mechanism that seeks to model specific aromatic species and therefore
separates a particular aromatic species (e.g., toluene) from the lumped aromatics category. This can change the average reactivity of the “lumped” species, because the reactivity of toluene may be different than that of the xylenes and ethyl benzene, which toluene may have been lumped with. This means that to update the toluene chemistry, the entire mechanism has to be updated (Whitten et al., 2010). Since even modest changes made to condensed mechanisms require that the entire mechanism be reevaluated, the development of condensed mechanisms suitable for specialized applications such as wildland or WUI fires has been limited. This difficulty in updating the chemical lumping is not limited to the chemical mechanisms.
A third issue is that condensed mechanisms have been designed with gas-phase chemistry in mind, without consideration for partitioning between the gas phase and clouds or aerosols. In some cases, species are “lumped” together across a wide range of Henry’s law constants, which makes it difficult to incorporate cloud and multiphase aerosol chemistry into the models, to predict the concentrations of water-soluble toxicants (e.g., isocyanic acid) and to predict deposition. This limitation affects the prediction of SOA and could affect the accuracy of gas-phase chemistry prediction, especially in the near field, by making it difficult to accurately account for the loss of reactive water-soluble gas-phase species to clouds and wet aerosols (e.g., formaldehyde, formic acid, HONO, HCl).
A fourth issue is the omission of classes of compounds (e.g., Coggon et al., 2019). For example, condensed mechanisms are not designed to track IVOCs, which are emitted as particulate matter but volatilize in the near field. Nor do they typically track furans, which have been reported to account for roughly 10 percent of ozone formation in the first 4 hours of wildfire smoke aging (Coggon et al., 2019).
These limitations are of particular concern for wildland fire and WUI fire model applications, given the importance of tracking toxicants for exposure assessment and the abundance of water-soluble species and IVOCs emitted by WUI fires.
Finding: Current models lack the chemical specificity needed to track in detail the types of toxicants associated with wildland fires and WUI fires.
Research need: Development of condensed chemical mechanisms is needed for use in applying chemical transport models to WUI fires.
Sub-Grid-Cell Chemical Processing of Emissions
For industrial point sources, near-source limitations of chemical transport models are frequently overcome through the use of sub-grid-cell (i.e., plume-in-grid [PiG]) chemical modeling tools; however, such tools are not designed for wildland fire or WUI fire emissions. Chemical transport modeling (e.g., CMAQ; Appel et al., 2021) is typically performed using gridded domains with a horizontal resolution of 12 km, 36 km, or more. The vertical depth of the model grid cells increases with height above ground level, and ranges from depths as small as tens of meters to depths as large as hundreds of meters at higher elevations. When emissions are modeled, they are assumed to immediately and completely mix throughout the grid cell into which they are emitted. Thus, the horizontal extent of the grid cell means that spatial variability in emissions over roughly kilometer scales is lost. Since fire burn fronts are often at smaller spatial scales (Freitas et al., 2007; Sokolik et al., 2019), chemical transport models will unrealistically mix fire plumes.
For sources with sub-grid scales, the unrealistic, immediate dilution of emissions leads chemical transport models to underestimate the rates of reactions. The rate of a chemical reaction scales directly with concentration, and therefore dilution of an emission that might be occurring over a 1 km2 area to a grid cell that is 36 km by 36 km in extent will reduce concentrations of the emitted species by a factor of more than 1,000. When the reactants are mixed over a large volume in a simulation, the reactions also are simulated to occur over a large volume. For unimolecular chemical reactions, using a large grid-cell size may have only a modest effect on accuracy. Many atmospheric chemical reactions are bimolecular, however. Emission plumes, occurring over a 1 km2 area, with multiple reactants participating in bimolecular reactions, that are immediately mixed over an area of 1,000 km2, will have the rates of the bimolecular reactions reduced by a factor of 106. Thus, even if these reactions are assumed to occur over a volume that is a factor of 103 larger, the volume-integrated rate of reaction will still be orders of magnitude too low. Thus, modeling of chemical transformations at sub-grid-cell levels is frequently needed.
The usefulness of sub-grid-cell processing is well known, and tools that model sub-grid-cell plumes within the context of a gridded model (PiG modeling tools) have been developed (Karamchandani et al., 2011; Wei et al., 2021). These PiG tools have their own reduced chemical mechanisms, and those mechanisms have been developed primarily for modeling industrial plumes, especially plumes of power plants that are rich in NOx (Karamchandani et al., 2011). The PiG tools designed to model industrial point sources model plume mixing with the surrounding atmosphere based on an industrial point-source plume structure, which differs from the typical wildland fire or WUI fire plume structure. Because the chemistry and plume dispersion currently available within PiG tools are not well suited for fire plumes, some wildland fire modeling studies have made use of box models to perform initial sub-grid-scale chemistry (Peng et al., 2021). However, it should be noted that the general framework of PiG tools could be useful for fire modeling.
Finding: Current models lack detailed physical modeling of the unique features of fire plume structures.
Finding: Models fail to capture key chemical interactions within the plume, including the interactions of emission sources with ambient air, clouds, and aerosols.
Research need: Development of sub-grid-cell processing techniques suitable for fire plumes is needed.
Research need: Regional chemical transport models need substantial enhancements in order to adequately describe WUI fire plumes, and research support is needed to drive these improvements.
Overall, chemical transport models, when used in combination with field measurements, are powerful tools for understanding and predicting air quality impacts of fires. Enhancements to chemical transport models, coupled with measurements to evaluate the performance of the improvements, could be used to develop new capabilities, including but not limited to the following:
- Modeling of individual chemical species that are either toxicants or surrogates for the emission of classes of WUI emissions, with consideration for properties that affect partitioning between the gas phase, clouds, and aerosols (e.g., Henry’s law constant, vapor pressure)
- Development of sub-grid-cell models that could be used to model the mixing of fire plumes with ambient air and the structure of single-structure or neighborhood-scale plumes
- Development of chemical transport models that could be used in prognostic mode to facilitate first-responder activity during environmental crises, and communication/decision-making coordination (a technology link to communicate predictions at the different scales of responders)
Research need: A combination of research approaches is needed to understand and predict toxicant concentrations downwind of WUI fires and ultimately mitigate their health risks.
Research need: There is a need to identify a subset of WUI fire–associated species (based on their toxicity or importance in chemistry) that are prioritized in both measurement and modeling efforts; attention is needed toward selecting priority species that span a range of physicochemical properties (e.g., vapor pressure, Henry’s law constant, oxidation state) and reactivity.
REFERENCES
Ahern, A. T., L. Goldberger, L. Jahl, J. Thornton, and R. C. Sullivan. 2018. “Production of N2O5 and ClNO2 through Nocturnal Processing of Biomass-Burning Aerosol.” Environmental Science & Technology 52(2):550–559. https://doi.org/10.1021/acs.est.7b04386.
Ahmadov, R., G. Grell, E. James, I. Csiszar, M. Tsidulko, B. Pierce, et al. 2017. “Using VIIRS Fire Radiative Power Data to Simulate Biomass Burning Emissions, Plume Rise and Smoke Transport in a Real-Time Air Quality Modeling System.” 2017. IEEE International Geoscience and Remote Sensing Symposium 2806–2808. New York, NY: IEEE.
Akagi, S. K., R. J. Yokelson, C. Wiedinmyer, M. J. Alvarado, J. S. Reid, T. Karl, J. D. Crounse, and P. O. Wennberg. 2011. “Emission Factors for Open and Domestic Biomass Burning for Use in Atmospheric Models.” Atmospheric Chemistry and Physics 11(9):4039–4072. https://doi.org/10.5194/acp-11-4039-2011.
Akherati, A., Y. He, M. M. Coggon, A. R. Koss, A. L. Hodshire, K. Sekimoto, C. Warneke, J. de Gouw, L. Yee, J. H. Seinfeld, T. B. Onasch, S. C. Herndon, W. B. Knighton, C. D. Cappa, M. J. Kleeman, C. Y. Lim, J. H. Kroll, J. R. Pierce, and S. H. Jathar. 2020. “Oxygenated Aromatic Compounds are Important Precursors of Secondary Organic Aerosol in Biomass-Burning Emissions.” Environmental Science & Technology 54(14):8568–8579. https://doi.org/10.1021/acs.est.0c01345.
Alvarado, M. J., J. A. Logan, J. Mao, E. Apel, D. Riemer, D. Blake, R. C. Cohen, K. E. Min, A. E. Perring, E. C. Browne, P. J. Wooldridge, G. S. Diskin, G. W. Sachse, H. Fuelberg, W. R. Sessions, D. L. Harrigan, G. Huey, J. Liao, A. Case-Hanks, J. L. Jimenez, M. J. Cubison, S. A. Vay, A. J. Weinheimer, D. J. Knapp, D. D. Montzka, F. M. Flocke, I. B. Pollack, P. O. Wennberg, A. Kurten, J. Crounse, J. M. S. Clair, A. Wisthaler, T. Mikoviny, R. M. Yantosca, C. C. Carouge, and P. Le Sager. 2010. “Nitrogen Oxides and PAN in Plumes from Boreal Fires during ARCTAS-B and Their Impact on Ozone: An Integrated Analysis of Aircraft and Satellite Observations.” Atmospheric Chemistry and Physics 10(20):9739–9760. https://doi.org/10.5194/acp-10-9739-2010.
Alves, C. A., A. Vicente, C. Monteiro, C. Goncalves, M. Evtyugina, and C. Pio. 2011. “Emission of Trace Gases and Organic Components in Smoke Particles from a Wildfire in a Mixed-Evergreen Forest in Portugal.” Science of the Total Environment 409(8):1466–1475. https://doi.org/10.1016/j.scitotenv.2010.12.025.
Andreae, M. O. 2019. “Emission of Trace Gases and Aerosols from Biomass Burning – An Updated Assessment.” Atmospheric Chemistry and Physics 19(13):8523–8546. https://doi.org/10.5194/acp-19-8523-2019.
Appel, K. W., O. Bash, K. M. Fahey, M. Foley, R. C. Gilliam, C. Hogrefe, . . . and D. C. Wong. 2021. “The Community Multiscale Air Quality (CMAQ) Model Versions 5.3 and 5.3.1: System Updates and Evaluation.” Geoscientific Model Development 14(5):2867–2897.
Arakaki, T., C. Anastasio, Y. Kuroki, H. Nakajima, K. Okada, Y. Kotani, D. Handa, S. Azechi, T. Kimura, A. Tsuhako, and Y. Miyagi. 2013. “A General Scavenging Rate Constant for Reaction of Hydroxyl Radical with Organic Carbon in Atmospheric Waters.” Environmental Science & Technology 47(15):8196–8203. https://doi.org/10.1021/es401927b.
Barth, M. C., B. Ervens, H. Herrmann, A. Tilgner, V. F. McNeill, W. G. Tsui, L. Deguillaume, N. Chaumerliac, A. Carlton, and S. M. Lance. 2021. “Box Model Intercomparison of Cloud Chemistry.” Journal of Geophysical Research: Atmospheres 126(21):e2021JD035486. https://doi.org/10.1029/2021JD035486.
Bianco, A., M. Passananti, M. Brigante, and G. Mailhot. 2020. “Photochemistry of the Cloud Aqueous Phase: A Review.” Molecules 25(2):423. https://doi.org/10.3390/molecules25020423.
Blando, J. D., and B. J. Turpin. 2000. “Secondary Organic Aerosol Formation in Cloud and Fog Droplets: A Literature Evaluation of Plausibility.” Atmospheric Environment 34(10):1623–1632. https://doi.org/10.1016/S1352-2310(99)00392-1.
Brégonzio-Rozier, L., C. Giorio, F. Siekmann, E. Pangui, S. B. Morales, B. Temime-Roussel, A. Gratien, V. Michoud, M. Cazaunau, H. L. DeWitt, A. Tapparo, A. Monod, and J. F. Doussin. 2016. “Secondary Organic Aerosol Formation from Isoprene Photooxidation during Cloud Condensation–Evaporation Cycles.” Atmospheric Chemistry and Physics 16(3):1747–1760. https://doi.org/10.5194/acp-16-1747-2016.
Briggs, N. L., D. A. Jaffe, H. Gao, J. R. Hee, P. M. Baylon, Q. Zhang, S. Zhou, S. C. Collier, P. D. Sampson, and R. A. Cary. 2017. “Particulate Matter, Ozone, and Nitrogen Species in Aged Wildfire Plumes Observed at the Mount Bachelor Observatory.” Aerosol and Air Quality Research 16(12):3075–3087. https://doi.org/10.4209/aaqr.2016.03.0120.
Budisulistiorini, S. H., M. R. Canagaratna, P. L. Croteau, W. J. Marth, K. Baumann, E. S. Edgerton, S. L. Shaw, E. M. Knipping, D. R. Worsnop, J. T. Jayne, A. Gold, and J. D. Surratt. 2013. “Real-Time Continuous Characterization of Secondary Organic Aerosol Derived from Isoprene Epoxydiols in Downtown Atlanta, Georgia, Using the Aerodyne Aerosol Chemical Speciation Monitor.” Environmental Science & Technology 47(11):5686–5694. https://doi.org/10.1021/es400023n.
Carlton, A. G., B. J. Turpin, H. J. Lim, K. E. Altieri, and S. Seitzinger. 2006. “Link between Isoprene and Secondary Organic Aerosol (SOA): Pyruvic Acid Oxidation Yields Low Volatility Organic Acids in Clouds.” Geophysical Research Letters 33(6). https://doi.org/10.1029/2005GL025374.
Chan, A. W. H., K. Kautzman, P. Chhabra, J. Surratt, M. Chan, J. Crounse, A. Kürten, P. Wennberg, R. Flagan, and J. Seinfeld. 2009. “Secondary Organic Aerosol Formation from Photooxidation of Naphthalene and Alkylnaphthalenes: Implications for Oxidation of Intermediate Volatility Organic Compounds (IVOCs).” Atmospheric Chemistry and Physics 9(9): 3049–3060. https://doi.org/10.5194/acp-9-3049-2009.
Chang, J. L., and J. E. Thompson. 2010. “Characterization of Colored Products Formed during Irradiation of Aqueous Solutions Containing H2O2 and Phenolic Compounds.” Atmospheric Environment 44(4):541–551. https://doi.org/10.1016/j.atmosenv.2009.10.042.
Charbouillot, T., S. Gorini, G. Voyard, M. Parazols, M. Brigante, L. Deguillaume, A.-M. Delort, and G. Mailhot. 2012. “Mechanism of Carboxylic Acid Photooxidation in Atmospheric Aqueous Phase: Formation, Fate and Reactivity.” Atmospheric Environment 56:1–8. https://doi.org/10.1016/j.atmosenv.2012.03.079.
Cheng, M. D. 2018. “Atmospheric Chemistry of Hydrogen Fluoride.” Journal of Atmospheric Chemistry 75:1–16. https://doi.org/10.1007/s10874-017-9359-7.
Chow, J. C., J. G. Watson, L.-W.A. Chen, M. Chang, N. F. Robinson, D. L. Trimble, and S. D. Kohl. 2007. “The IMPROVE: A temperature protocol for thermal/optical carbon analysis: Maintaining consistency with a long-term database.” Journal of the Air & Waste Management Association 57(9):1014–1023. https://doi.org/10.3155/1047-3289.57.9.1014.
Ciobanu, V. G., C. Marcolli, U. K. Krieger, U. Weers, and T. Peter. 2009. “Liquid–Liquid Phase Separation in Mixed Organic/Inorganic Aerosol Particles.” The Journal of Physical Chemistry A 113(41):10966–10978. https://doi.org/10.1021/jp905054d.
Coggon, M. M., C. Y. Lim, A. R. Koss, K. Sekimoto, B. Yuan, J. B. Gilman, D. H. Hagan, V. Selimovic, K. J. Zarzana, S. S. Brown, J. M. Roberts, M. Müller, R. Yokelson, A. Wisthaler, J. E. Krechmer, J. L. Jimenez, C. Cappa, J. H. Kroll, J. de Gouw, and C. Warneke. 2019. “OH Chemistry of Non-methane Organic Gases (NMOGs) Emitted from Laboratory and Ambient Biomass Burning Smoke: Evaluating the Influence of Furans and Oxygenated Aromatics on Ozone and Secondary NMOG Formation.” Atmospheric Chemistry and Physics 19(23):14875–14899. https://doi.org/10.5194/acp-19-14875-2019.
Cook, R. D., Y.-H. Lin, Z. Peng, E. Boone, R. K. Chu, J. E. Dukett, M. J. Gunsch, W. Zhang, N. Tolic, and A. Laskin. 2017. “Biogenic, Urban, and Wildfire Influences on the Molecular Composition of Dissolved Organic Compounds in Cloud Water.” Atmospheric Chemistry and Physics 17(24):15167–15180. https://doi.org/10.5194/acp-17-15167-2017.
Decker, Z. C. J., K. J. Zarzana, M. Coggon, K. E. Min, I. Pollack, T. B. Ryerson, J. Peischl, P. Edwards, W. P. Dube, M. Z. Markovic, J. M. Roberts, P. R. Veres, M. Graus, C. Warneke, J. de Gouw, L. E. Hatch, K. C. Barsanti, and S. S. Brown. 2019. “Nighttime Chemical Transformation in Biomass Burning Plumes: A Box Model Analysis Initialized with Aircraft Observations.” Environmental Science & Technology 53(5):2529–2538. https://doi.org/10.1021/acs.est.8b05359.
Decker, Z. C., M. A. Robinson, K. C. Barsanti, I. Bourgeois, M. M. Coggon, J. P. DiGangi, G. S. Diskin, F. M. Flocke, A. Franchin, and C. D. Fredrickson. 2021. “Nighttime and Daytime Dark Oxidation Chemistry in Wildfire Plumes: An Observation and Model Analysis of FIREX-AQ Aircraft Data.” Atmospheric Chemistry and Physics 21(21):16293–16317. https://doi.org/10.5194/acp-21-16293-2021.
Dreessen, J., J. Sullivan, and R. Delgado. 2016. “Observations and Impacts of Transported Canadian Wildfire Smoke on Ozone and Aerosol Air Quality in the Maryland Region on June 9–12, 2015.” Journal of the Air & Waste Management Association 66(9):842–862. https://doi.org/10.1080/10962247.2016.1161674.
Ellis, D. A., J. W. Martin, S. A. Mabury, M. D. Hurley, M. P. Sulbaek Andersen, and T. J. Wallington. 2003. “Atmospheric Lifetime of Fluorotelomer Alcohols.” Environmental Science & Technology 37(17):3816–3820. https://doi.org/10.1021/es034136j.
Ellis, D. A., J. W. Martin, A. O. De Silva, S. A. Mabury, M. D. Hurley, M. P. Sulbaek Andersen, and T. J. Wallington. 2004. “Degradation of Fluorotelomer Alcohols: A Likely Atmospheric Source of Perfluorinated Carboxylic Acids.” Environmental Science & Technology 38(12):3316–3321. https://doi.org/10.1021/es049860w.
Enami, S., Y. Sakamoto, and A. J. Colussi. 2014. “Fenton Chemistry at Aqueous Interfaces.” Proceedings of the National Academy of Sciences 111(2):623–628. https://doi.org/10.1073/pnas.1314885111.
Evtyugina, M., A. I. Calvo, T. Nunes, C. Alves, A. P. Fernandes, L. Tarelho, . . . and C. Pio. 2013. “VOC Emissions of Smouldering Combustion from Mediterranean Wildfires in Central Portugal.” Atmospheric Environment 64:339–348.
Fann, N., B. Alman, R. A. Broome, G. G. Morgan, F. H. Johnston, G. Pouliot, and A. G. Rappold. 2018. “The Health Impacts and Economic Value of Wildland Fire Episodes in the U.S.: 2008–2012.” Science of the Total Environment 610–611:802–809. https://doi.org/10.1016/j.scitotenv.2017.08.024.
Faxon, C. B., and D. T. Allen. 2013. “Chlorine Chemistry in Urban Atmospheres: A Review.” Environmental Chemistry 10(3):221–233. https://doi.org/10.1071/EN13026.
Freitas, S. R., K. M. Longo, R. Chatfield, D. Latham, M. A. F. Silva Dias, M. O. Andreae, . . . and J. A. Carvalho Jr. 2007. “Including the Sub-grid Scale Plume Rise of Vegetation Fires in Low Resolution Atmospheric Transport Models.” Atmospheric Chemistry and Physics 7(13):3385–3398.
Friedli, H., E. Atlas, V. Stroud, L. Giovanni, T. Campos, and L. Radke. 2001. “Volatile Organic Trace Gases Emitted from North American Wildfires.” Global Biogeochemical Cycles 15(2):435–452. https://doi.org/10.1029/2000GB001328.
Garofalo, L. A., M. A. Pothier, E. J. Levin, T. Campos, S. M. Kreidenweis, and D. K. Farmer. 2019. “Emission and Evolution of Submicron Organic Aerosol in Smoke from Wildfires in the Western United States.” ACS Earth and Space Chemistry 3(7):1237–1247. https://doi.org/10.1021/acsearthspacechem.9b00125.
Gilardoni, S., P. Massoli, M. Paglione, L. Giulianelli, C. Carbone, M. Rinaldi, S. Decesari, S. Sandrini, F. Costabile, G. P. Gobbi, M. C. Pietrogrande, M. Visentin, F. Scotto, S. Fuzzi, and M. C. Facchini. 2016. “Direct Observation of Aqueous Secondary Organic Aerosol from Biomass-Burning Emissions.” Proceedings of the National Academy of Sciences 113(36):10013–10018. https://doi.org/10.1073/pnas.1602212113.
Gilman, J., B. Lerner, W. Kuster, P. D. Goldan, C. Warneke, P. Veres, J. Roberts, J. De Gouw, I. Burling, and R. Yokelson. 2015. “Biomass Burning Emissions and Potential Air Quality Impacts of Volatile Organic Compounds and Other Trace Gases from Fuels Common in the US.” Atmospheric Chemistry and Physics 15(24):13915–13938. https://doi.org/10.5194/acpd-15-21713-2015.
Goldberger, L. A., L. G. Jahl, J. A. Thornton, and R. C. Sullivan. 2019. “N2O5 Reactive Uptake Kinetics and Chlorine Activation on Authentic Biomass-Burning Aerosol.” Environmental Science: Processes & Impacts 21(10):1684–1698. https://doi.org/10.1039/c9em00330d.
Gong, X., A. S. Kaulfus, U. S. Nair, and D. A. Jaffe. 2017. “Quantifying O3 Impacts in Urban Areas Due to Wildfires Using a Generalized Additive Model.” Environmental Science & Technology 51(22):13216–13223. https://doi.org/10.1021/acs.est.7b03130.
Goodrick, S. L., G. L. Achtemeier, N. K. Larkin, Y. Liu, and T. Strand. 2012. “Modelling Smoke Transport from Wildland Fires: A Review.” International Journal of Wildland Fire 22(1):83–94.
Hall, S. M., S. Patton, M. Petreas, S. Zhang, A. L. Phillips, K. Hoffman, and H. M. Stapleton. 2020. “Per- and Polyfluoroalkyl Substances in Dust Collected from Residential Homes and Fire Stations in North America.” Environmental Science & Technology 54(22):14558–14567. https://doi.org/10.1021/acs.est.0c04869.
Harrison, M. A. J., S. Barra, D. Borghesi, D. Vione, C. Arsene, and R. Iulian Olariu. 2005. “Nitrated Phenols in the Atmosphere: A Review.” Atmospheric Environment 39(2):231–248. https://doi.org/10.1016/j.atmosenv.2004.09.044.
Hatch, L. E., W. Luo, J. F. Pankow, R. J. Yokelson, C. E. Stockwell, and K. Barsanti. 2015. “Identification and Quantification of Gaseous Organic Compounds Emitted from Biomass Burning Using Two-Dimensional Gas Chromatography–Time-of-Flight Mass Spectrometry.” Atmospheric Chemistry and Physics 15(4):1865–1899. https://doi.org/10.5194/acp-15-1865-2015.
Hatch, L. E., R. J. Yokelson, C. E. Stockwell, P. R. Veres, I. J. Simpson, D. R. Blake, J. J. Orlando, and K. C. Barsanti. 2017. “Multi-instrument Comparison and Compilation of Non-methane Organic Gas Emissions from Biomass Burning and Implications for Smoke-Derived Secondary Organic Aerosol Precursors.” Atmospheric Chemistry and Physics 17(2):1471–1489. https://doi.org/10.5194/acp-17-1471-2017.
Hatch, L. E., A. Rivas-Ubach, C. N. Jen, M. Lipton, A. H. Goldstein, and K. C. Barsanti. 2018. “Measurements of I/SVOCs in Biomass-Burning Smoke Using Solid-Phase Extraction Disks and Two-Dimensional Gas Chromatography.” Atmospheric Chemistry and Physics 18(24):17801–17817. https://doi.org/10.5194/acp-18-17801-2018.
Hawkins, L. N., M. J. Baril, N. Sedehi, M. M. Galloway, D. O. De Haan, G. P. Schill, and M. A. Tolbert. 2014. “Formation of Semisolid, Oligomerized Aqueous SOA: Lab Simulations of Cloud Processing.” Environmental Science & Technology 48(4):2273–2280. https://doi.org/10.1021/es4049626.
Hobbs, P. V., P. Sinha, R. J. Yokelson, T. J. Christian, D. R. Blake, S. Gao, T. W. Kirchstetter, T. Novakov, and P. Pilewskie. 2003. “Evolution of Gases and Particles from a Savanna Fire in South Africa.” Journal of Geophysical Research: Atmospheres 108(D13). https://doi.org/10.1029/2002JD002352.
Hodshire, A. L., A. Akherati, M. J. Alvarado, B. Brown-Steiner, S. H. Jathar, J. L. Jimenez, S. M. Kreidenweis, C. R. Lonsdale, T. B. Onasch, A. M. Ortega, and J. R. Pierce. 2019. “Aging Effects on Biomass Burning Aerosol Mass and Composition: A Critical Review of Field and Laboratory Studies.” Environmental Science & Technology 53(17):10007–10022. https://doi.org/10.1021/acs.est.9b02588.
Huang, R., R. Lal, M. Qin, Y. Hu, A. G. Russell, M. T. Odman, S. Afrin, F. Garcia-Menendez, and S. M. O’Neill. 2021. “Application and Evaluation of a Low-Cost PM Sensor and Data Fusion with CMAQ Simulations to Quantify the Impacts of Prescribed Burning on Air Quality in Southwestern Georgia, USA.” Journal of the Air & Waste Management Association 71(7):815–829. https://doi.org/10.1080/10962247.2021.1924311.
Jaffe, D. A., and N. L. Wigder. 2012. “Ozone Production from Wildfires: A Critical Review.” Atmospheric Environment 51:1–10. https://doi.org/10.1016/j.atmosenv.2011.11.063.
Jaffe, D. A., S. M. O’Neill, N. K. Larkin, A. L. Holder, D. L. Peterson, J. E. Halofsky, and A. G. Rappold. 2020. “Wildfire and Prescribed Burning Impacts on Air Quality in the United States.” Journal of the Air & Waste Management Association 70(6):583–615. https://doi.org/10.1080/10962247.2020.1749731.
Jiang, X., and E. H. Enki Yoo. 2019. “Modeling Wildland Fire-Specific PM2.5 Concentrations for Uncertainty-Aware Health Impact Assessments.” Environmental Science & Technology 53(20):11828–11839. https://doi.org/10.1021/acs.est.9b02660.
Juncosa Calahorrano, J. F., J. Lindaas, K. O’Dell, B. B. Palm, Q. Peng, F. Flocke, I. B. Pollack, L. A. Garofalo, D. K. Farmer, and J. R. Pierce. 2021. “Daytime Oxidized Reactive Nitrogen Partitioning in Western US Wildfire Smoke Plumes.” Journal of Geophysical Research: Atmospheres 126(4):e2020JD033484. https://doi.org/10.1029/2020JD033484.
Junghenn Noyes, K., R. Kahn, A. Sedlacek, L. Kleinman, J. Limbacher, and Z. Li. 2020. “Wildfire Smoke Particle Properties and Evolution, from Space-Based Multi-Angle Imaging.” Remote Sensing 12(5):769. https://doi.org/10.3390/rs12050769.
Karamchandani, P., K. Vijayaraghavan, and G. Yarwood. 2011. “Sub-grid Scale Plume Modeling.” Atmosphere 2(3):389–406.
Koss, A. R., K. Sekimoto, J. B. Gilman, V. Selimovic, M. M. Coggon, K. J. Zarzana, B. Yuan, B. M. Lerner, S. S. Brown, and J. L. Jimenez. 2018. “Non-methane Organic Gas Emissions from Biomass Burning: Identification, Quantification, and Emission Factors from PTR-ToF during the FIREX 2016 Laboratory Experiment.” Atmospheric Chemistry and Physics 18(5):3299–3319. https://doi.org/10.5194/acp-18-3299-2018.
Kroll, J. H., and J. H. Seinfeld. 2008. “Chemistry of Secondary Organic Aerosol: Formation and Evolution of Low-Volatility Organics in the Atmosphere.” Atmospheric Environment 42(16):3593–3624. https://doi.org/10.1016/j.atmosenv.2008.01.003.
Lambe, A. T., T. B. Onasch, D. R. Croasdale, J. P. Wright, A. T. Martin, J. P. Franklin, P. Massoli, J. H. Kroll, M. R. Canagaratna, W. H. Brune, D. R. Worsnop, and P. Davidovits. 2012. “Transitions from Functionalization to Fragmentation Reactions of Laboratory Secondary Organic Aerosol (SOA) Generated from the OH Oxidation of Alkane Precursors.” Environmental Science & Technology 46(10):5430–5437. https://doi.org/10.1021/es300274t.
Lamkaddam, H., J. Dommen, A. Ranjithkumar, H. Gordon, G. Wehrle, J. Krechmer, F. Majluf, D. Salionov, J. Schmale, S. Bjelić, K. S. Carslaw, I. E. Haddad, and U. Baltensperger. 2021. “Large Contribution to Secondary Organic Aerosol from Isoprene Cloud Chemistry.” Science Advances 7(13):eabe2952. https://doi.org/10.1126/sciadv.abe2952.
Lee, A. K. Y., P. Herckes, W. R. Leaitch, A. M. Macdonald, and J. P. D. Abbatt. 2011. “Aqueous OH Oxidation of Ambient Organic Aerosol and Cloud Water Organics: Formation of Highly Oxidized Products.” Geophysical Research Letters 38(11). https://doi.org/10.1029/2011GL047439.
Leslie, M. D., M. Ridoli, J. G. Murphy, and N. Borduas-Dedekind. 2019. “Isocyanic Acid (HNCO) and Its Fate in the Atmosphere: A Review.” Environmental Science: Processes & Impacts 21(5):793–808. https://doi.org/10.1039/c9em00003h.
Li, W., L. Liu, J. Zhang, L. Xu, Y. Wang, Y. Sun, and Z. Shi. 2021. “Microscopic Evidence for Phase Separation of Organic Species and Inorganic Salts in Fine Ambient Aerosol Particles.” Environmental Science & Technology 55(4):2234–2242. https://doi.org/10.1021/acs.est.0c02333.
Li, Y. J., D. D. Huang, H. Y. Cheung, A. K. Y. Lee, and C. K. Chan. 2014. “Aqueous-Phase Photochemical Oxidation and Direct Photolysis of Vanillin – A Model Compound of Methoxy Phenols from Biomass Burning.” Atmospheric Chemistry and Physics 14(6):2871–2885. https://doi.org/10.5194/acp-14-2871-2014.
Lim, Y. B., Y. Tan, M. J. Perri, S. P. Seitzinger, and B. J. Turpin. 2010. “Aqueous Chemistry and Its Role in Secondary Organic Aerosol (SOA) Formation.” Atmospheric Chemistry and Physics 10(21):10521–10539. https://doi.org/10.5194/acp-10-10521-2010.
Lim, Y., Y. Tan, and B. Turpin. 2013. “Chemical Insights, Explicit Chemistry, and Yields of Secondary Organic Aerosol from OH Radical Oxidation of Methylglyoxal and Glyoxal in the Aqueous Phase.” Atmospheric Chemistry and Physics 13(17):8651–8667. https://doi.org/10.5194/acp-13-8651-2013.
Lin, G., S. Sillman, J. Penner, and A. Ito. 2014. “Global Modeling of SOA: The Use of Different Mechanisms for Aqueous-Phase Formation.” Atmospheric Chemistry and Physics 14(11):5451–5475. https://doi.org/10.5194/acp-14-5451-2014.
Lin, P., G. Engling, and J. Yu. 2010. “Humic-Like Substances in Fresh Emissions of Rice Straw Burning and in Ambient Aerosols in the Pearl River Delta Region, China.” Atmospheric Chemistry and Physics 10(14):6487–6500. https://doi.org/10.5194/acp-10-6487-2010.
Liu, J., L. W. Horowitz, S. Fan, A. G. Carlton, and H. Levy. 2012. “Global In-Cloud Production of Secondary Organic Aerosols: Implementation of a Detailed Chemical Mechanism in the GFDL Atmospheric Model AM3.” Journal of Geophysical Research: Atmospheres 117(D15). https://doi.org/10.1029/2012JD017838.
Liu, Y., J. Liggio, T. Harner, L. Jantunen, M. Shoeib, and S. M. Li. 2014. “Heterogeneous OH Initiated Oxidation: A Possible Explanation for the Persistence of Organophosphate Flame Retardants in Air.” Environmental Science & Technology 48(2):1041–1048. https://doi.org/10.1021/es404515k.
Loeffler, K. W., C. A. Koehler, N. M. Paul, and D. O. De Haan. 2006. “Oligomer Formation in Evaporating Aqueous Glyoxal and Methyl Glyoxal Solutions.” Environmental Science & Technology 40(20):6318–6323. https://doi.org/10.1021/es060810w.
Lu, X., L. Zhang, X. Yue, J. Zhang, D. A. Jaffe, A. Stohl, Y. Zhao, and J. Shao. 2016. “Wildfire Influences on the Variability and Trend of Summer Surface Ozone in the Mountainous Western United States.” Atmospheric Chemistry and Physics 16(22):14687–14702. https://doi.org/10.5194/acp-16-14687-2016.
Luecken, D. J., S. L. Napelenok, M. Strum, R. Scheffe, and S. Phillips. 2018. “Sensitivity of Ambient Atmospheric Formaldehyde and Ozone to Precursor Species and Source Types across the United States.” Environmental Science & Technology 52(8):4668–4675. https://doi.org/10.1021/acs.est.7b05509.
McFall, A. S., A. W. Johnson, and C. Anastasio. 2020. “Air–Water Partitioning of Biomass-Burning Phenols and the Effects of Temperature and Salinity.” Environmental Science & Technology 54(7):3823–3830. https://doi.org/10.1021/acs.est.9b06443.
McMeeking, G. R., S. M. Kreidenweis, S. Baker, C. M. Carrico, J. C. Chow, J. L. Collett Jr, W. M. Hao, A. S. Holden, T. W. Kirchstetter, and W. C. Malm. 2009. “Emissions of Trace Gases and Aerosols during the Open Combustion of Biomass in the Laboratory.” Journal of Geophysical Research: Atmospheres 114(D19). https://doi.org/10.1029/2009JD011836.
McNeill, V. F. 2015. “Aqueous Organic Chemistry in the Atmosphere: Sources and Chemical Processing of Organic Aerosols.” Environmental Science & Technology 49(3):1237–1244. https://doi.org/10.1021/es5043707.
Ng, N. L., S. S. Brown, A. T. Archibald, E. Atlas, R. C. Cohen, J. N. Crowley, D. A. Day, N. M. Donahue, J. L. Fry, H. Fuchs, R. J. Griffin, M. I. Guzman, H. Herrmann, A. Hodzic, Y. Iinuma, J. L. Jimenez, A. Kiendler-Scharr, B. H. Lee, D. J. Luecken, J. Mao, R. McLaren, A. Mutzel, H. D. Osthoff, B. Ouyang, B. Picquet-Varrault, U. Platt, H. O. T. Pye, Y. Rudich, R. H. Schwantes, M. Shiraiwa, J. Stutz, J. A. Thornton, A. Tilgner, B. J. Williams, and R. A. Zaveri. 2017. “Nitrate Radicals and Biogenic Volatile Organic Compounds: Oxidation, Mechanisms, and Organic Aerosol.” Atmospheric Chemistry and Physics 17(3):2103–2162. https://doi.org/10.5194/acp-17-2103-2017.
NOAA ARL (National Oceanic and Atmospheric Administration Air Resources Laboratory). 2012. The HYSPLIT. https://www.arl.noaa.gov/hysplit/ (accessed December 23, 2021).
NOAA ARL. 2021. HYSPLIT-based Smoke Forecasts. https://www.arl.noaa.gov/hysplit/smoke-forecasting/ (accessed October 28, 2021).
O’Dell, K., B. Ford, E. V. Fischer, and J. R. Pierce. 2019. “Contribution of Wildland-Fire Smoke to US PM2.5 and Its Influence on Recent Trends.” Environmental Science & Technology 53(4):1797–1804. https://doi.org/10.1021/acs.est.8b05430.
O’Dell, K., R. S. Hornbrook, W. Permar, E. J. T. Levin, L. A. Garofalo, E. C. Apel, N. J. Blake, A. Jarnot, M. A. Pothier, D. K. Farmer, L. Hu, T. Campos, B. Ford, J. R. Pierce, and E. V. Fischer. 2020. “Hazardous Air Pollutants in Fresh and Aged Western US Wildfire Smoke and Implications for Long-Term Exposure.” Environmental Science & Technology 54(19):11838–11847. https://doi.org/10.1021/acs.est.0c04497.
Ohura, T., A. Kitazawa, T. Amagai, and M. Makino. 2005. “Occurrence, Profiles, and Photostabilities of Chlorinated Polycyclic Aromatic Hydrocarbons Associated with Particulates in Urban Air.” Environmental Science & Technology 39(1):85–91. https://doi.org/10.1021/es040433s.
Ortiz-Montalvo, D. L., S. A. Häkkinen, A. N. Schwier, Y. B. Lim, V. F. McNeill, and B. J. Turpin. 2014. “Ammonium Addition (and Aerosol pH) Has a Dramatic Impact on the Volatility and Yield of Glyoxal Secondary Organic Aerosol.” Environmental Science & Technology 48(1):255–262. https://doi.org/10.1021/es4035667.
Paglione, M., S. Gilardoni, M. Rinaldi, S. Decesari, N. Zanca, S. Sandrini, L. Giulianelli, D. Bacco, S. Ferrari, and V. Poluzzi. 2020. “The Impact of Biomass Burning and Aqueous-Phase Processing on Air Quality: A Multi-year Source Apportionment Study in the Po Valley, Italy.” Atmospheric Chemistry and Physics 20(3):1233–1254. https://doi.org/10.5194/acp-20-1233-2020.
Palm, B. B., Q. Peng, C. D. Fredrickson, B. H. Lee, L. A. Garofalo, M. A. Pothier, S. M. Kreidenweis, D. K. Farmer, R. P. Pokhrel, Y. Shen, S. M. Murphy, W. Permar, L. Hu, T. L. Campos, S. R. Hall, K. Ullmann, X. Zhang, F. Flocke, E. V. Fischer, and J. A. Thornton. 2020. “Quantification of Organic Aerosol and Brown Carbon Evolution in Fresh Wildfire Plumes.” Proceedings of the National Academy of Sciences 117(47):29469–29477. https://doi.org/10.1073/pnas.2012218117.
Pang, H., Q. Zhang, X. Lu, K. Li, H. Chen, J. Chen, X. Yang, Y. Ma, J. Ma, and C. Huang. 2019. “Nitrite-Mediated Photooxidation of Vanillin in the Atmospheric Aqueous Phase.” Environmental Science & Technology 53(24):14253–14263. https://doi.org/10.1021/acs.est.9b03649.
Patoulias, D., E. Kallitsis, L. Posner, and S. N. Pandis. 2021. “Modeling Biomass Burning Organic Aerosol Atmospheric Evolution and Chemical Aging.” Atmosphere 12(12):1638. https://doi.org/10.3390/atmos12121638.
Paugam, R., M. Wooster, S. Freitas, and M. Val Martin. 2016. “A Review of Approaches to Estimate Wildfire Plume Injection Height within Large-Scale Atmospheric Chemical Transport Models.” Atmospheric Chemistry and Physics 16(2):907–925. https://doi.org/10.5194/acp-16-907-2016.
Paulson, S. E., P. J. Gallimore, X. M. Kuang, J. R. Chen, M. Kalberer, and D. H. Gonzalez. 2019. “A Light-Driven Burst of Hydroxyl Radicals Dominates Oxidation Chemistry in Newly Activated Cloud Droplets.” Science Advances 5(5):eaav7689. https://doi.org/10.1126/sciadv.aav7689.
Peng, Q., B. B. Palm, C. D. Fredrickson, B. H. Lee, S. R. Hall, K. Ullmann, . . . and J. Thornton. 2021. “Observations and Modeling of NOx Photochemistry and Fate in Fresh Wildfire Plumes.” ACS Earth and Space Chemistry 5(10):2652–2667.
Permar, W., Q. Wang, V. Selimovic, C. Wielgasz, R. J. Yokelson, R. S. Hornbrook, A. J. Hills, E. C. Apel, I.-T. Ku, Y. Zhou, B. C. Sive, A. P. Sullivan, J. L. Collett Jr, T. L. Campos, B. B. Palm, Q. Peng, J. A. Thornton, L. A. Garofalo, D. K. Farmer, S. M. Kreidenweis, E. J. T. Levin, P. J. DeMott, F. Flocke, E. V. Fischer, and L. Hu. 2021. “Emissions of Trace Organic Gases from Western U.S. Wildfires Based on WE-CAN Aircraft Measurements.” Journal of Geophysical Research: Atmospheres 126(11):e2020JD033838. https://doi.org/10.1029/2020JD033838.
Perri, M. J., S. Seitzinger, and B. J. Turpin. 2009. “Secondary Organic Aerosol Production from Aqueous Photooxidation of Glycolaldehyde: Laboratory Experiments.” Atmospheric Environment 43(8):1487–1497. https://doi.org/10.1016/j.atmosenv.2008.11.037.
Pike, K. A., P. L. Edmiston, J. J. Morrison, and J. A. Faust. 2021. “Correlation Analysis of Perfluoroalkyl Substances in Regional US Precipitation Events.” Water Research 190:116685. https://doi.org/10.1016/j.watres.2020.116685.
Posner, L. N., G. Theodoritsi, A. Robinson, G. Yarwood, B. Koo, R. Morris, M. Mavko, T. Moore, and S. N. Pandis. 2019. “Simulation of Fresh and Chemically-Aged Biomass Burning Organic Aerosol.” Atmospheric Environment 196:27–37. https://doi.org/10.1016/j.atmosenv.2018.09.055.
Real, E., K. S. Law, B. Weinzierl, M. Fiebig, A. Petzold, O. Wild, J. Methven, S. Arnold, A. Stohl, and H. Huntrieser. 2007. “Processes Influencing Ozone Levels in Alaskan Forest Fire Plumes during Long-Range Transport over the North Atlantic.” Journal of Geophysical Research: Atmospheres 112(D10). https://doi.org/10.1029/2006JD007576.
Reisch, M. S. 2018. “The Price of Fire Safety.” Chemical & Engineering News 97(2):16–19. https://www.firefightingfoam.com/assets/Uploads/ARTICLES-/jan14-19-Chemical-and-Engineering-News.pdf.
Riemer, D. D., E. C. Apel, J. J. Orlando, G. S. Tyndall, W. H. Brune, E. J. Williams, W. A. Lonneman, and J. D. Neece. 2008. “Unique Isoprene Oxidation Products Demonstrate Chlorine Atom Chemistry Occurs in the Houston, Texas Urban Area.” Journal of Atmospheric Chemistry 61(3):227–242. https://doi.org/10.1007/s10874-009-9134-5.
Riva, M., R. M. Healy, P. M. Flaud, E. Perraudin, J. C. Wenger, and E. Villenave. 2015. “Gas- and Particle-Phase Products from the Chlorine-Initiated Oxidation of Polycyclic Aromatic Hydrocarbons.” Journal of Physical Chemistry A 119(45): 11170–11181. https://doi.org/10.1021/acs.jpca.5b04610.
Roberts, J. M., and Y. Liu. 2019. “Solubility and Solution-Phase Chemistry of Isocyanic Acid, Methyl Isocyanate, and Cyanogen Halides.” Atmospheric Chemistry and Physics 19(7):4419–4437. https://doi.org/10.5194/acp-19-4419-2019.
Roberts, J. M., P. R. Veres, A. K. Cochran, C. Warneke, I. R. Burling, R. J. Yokelson, B. Lerner, J. B. Gilman, W. C. Kuster, R. Fall, and J. de Gouw. 2011. “Isocyanic Acid in the Atmosphere and Its Possible Link to Smoke-Related Health Effects.” Proceedings of the National Academy of Sciences 108(22):8966–8971. https://doi.org/10.1073/pnas.1103352108.
Robinson, A. L., N. M. Donahue, M. K. Shrivastava, E. A. Weitkamp, A. M. Sage, A. P. Grieshop, T. E. Lane, J. R. Pierce, and S. N. Pandis. 2007. “Rethinking Organic Aerosols: Semivolatile Emissions and Photochemical Aging.” Science 315(5816):1259–1262. https://doi.org/10.1126/science.1133061.
Rooney, B., Y. Wang, J. H. Jiang, B. Zhao, Z.-C. Zeng, and J. H. Seinfeld. 2020. “Air Quality Impact of the Northern California Camp Fire of November 2018.” Atmospheric Chemistry and Physics 20(23):14597–14616. https://doi.org/10.5194/acp-20-14597-2020.
Rossignol, S., K. Z. Aregahegn, L. Tinel, L. Fine, B. Noziere, and C. George. 2014. “Glyoxal Induced Atmospheric Photosensitized Chemistry Leading to Organic Aerosol Growth.” Environmental Science & Technology 48(6):3218–3227. https://doi.org/10.1021/es405581g.
Ruokojärvi, P., M. Aatamila, and J. Ruuskanen. 2000. “Toxic Chlorinated and Polyaromatic Hydrocarbons in Simulated House Fires.” Chemosphere 41(6):825–828. https://doi.org/10.1016/s0045-6535(99)00549-4.
Schauer, J. J., M. J. Kleeman, G. R. Cass, and B. R. Simoneit. 2001. “Measurement of Emissions from Air Pollution Sources. 3. C1–C29 Organic Compounds from Fireplace Combustion of Wood.” Environmental Science & Technology 35(9): 1716–1728. https://doi.org/10.1021/es001331e.
Seinfeld, J. H., and S. N. Pandis. 2016. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change. Hoboken, NJ: John Wiley & Sons.
Selimovic, V., R. J. Yokelson, C. Warneke, J. M. Roberts, J. d. Gouw, J. Reardon, and D. W. Griffith. 2018. “Aerosol Optical Properties and Trace Gas Emissions by PAX and OP-FTIR for Laboratory-Simulated Western US Wildfires during FIREX.” Atmospheric Chemistry and Physics 18(4):2929–2948. https://doi.org/10.5194/acp-18-2929-2018.
Sha, B., A. K. Dahlberg, K. Wiberg, and L. Ahrens. 2018. “Fluorotelomer Alcohols (FTOHs), Brominated Flame Retardants (BFRs), Organophosphorus Flame Retardants (OPFRs) and Cyclic Volatile Methylsiloxanes (cVMSs) in Indoor Air from Occupational and Home Environments.” Environmental Pollution 241:319–330. https://doi.org/10.1016/j.envpol.2018.04.032.
Shimizu, M. S., R. Mott, A. Potter, J. Zhou, K. Baumann, J. D. Surratt, B. Turpin, G. B. Avery, J. Harfmann, and R. J. Kieber. 2021. “Atmospheric Deposition and Annual Flux of Legacy Perfluoroalkyl Substances and Replacement Perfluoroalkyl Ether Carboxylic Acids in Wilmington, NC, USA.” Environmental Science & Technology Letters 8(5):366–372. https://doi.org/10.1021/acs.estlett.1c00251.
Sokolik, I. N., A. J. Soja, P. J. DeMott, and D. Winker. 2019. “Progress and Challenges in Quantifying Wildfire Smoke Emissions, Their Properties, Transport, and Atmospheric Impacts.” Journal of Geophysical Research: Atmospheres 124(23):13005–13025.
Spada, N. J., and N. P. Hyslop. 2018. “Comparison of Elemental and Organic Carbon Measurements between IMPROVE and CSN before and after Method Transitions.” Atmospheric Environment 178:1783-180. https://doi.org/10.1016/j.atmosenv.2018.01.043.
Stein, A., R. R. Draxler, G. D. Rolph, B. J. Stunder, M. Cohen, and F. Ngan. 2015. “NOAA’s HYSPLIT Atmospheric Transport and Dispersion Modeling System.” Bulletin of the American Meteorological Society 96(12):2059–2077. https://doi.org/10.1175/BAMS-D-14-00110.1.
Sun, Y. L., Q. Zhang, C. Anastasio, and J. Sun. 2010. “Insights into Secondary Organic Aerosol Formed via Aqueous-Phase Reactions of Phenolic Compounds Based on High Resolution Mass Spectrometry.” Atmospheric Chemistry and Physics 10(10):4809–4822. https://doi.org/10.5194/acp-10-4809-2010.
Surratt, J. D., A. W. H. Chan, N. C. Eddingsaas, M. Chan, C. L. Loza, A. J. Kwan, S. P. Hersey, R. C. Flagan, P. O. Wennberg, and J. H. Seinfeld. 2010. “Reactive Intermediates Revealed in Secondary Organic Aerosol Formation from Isoprene.” Proceedings of the National Academy of Sciences 107(15):6640–6645. https://doi.org/10.1073/pnas.0911114107.
Tabazadeh, A., R. J. Yokelson, H. Singh, P. V. Hobbs, J. Crawford, and L. Iraci. 2004. “Heterogeneous Chemistry Involving Methanol in Tropospheric Clouds.” Geophysical Research Letters 31(6). https://doi.org/10.1029/2003GL018775.
Tan, Y., Y. Lim, K. Altieri, S. Seitzinger, and B. Turpin. 2012. “Mechanisms Leading to Oligomers and SOA through Aqueous Photooxidation: Insights from OH Radical Oxidation of Acetic Acid and Methylglyoxal.” Atmospheric Chemistry and Physics 12(2):801–813. https://doi.org/10.5194/acp-12-801-2012.
Tanaka, P. L., D. D. Riemer, S. Chang, G. Yarwood, E. C. McDonald-Buller, E. C. Apel, J. J. Orlando, P. J. Silva, J. L. Jimenez, M. R. Canagaratna, J. D. Neece, C. B. Mullins, and D. T. Allen. 2003. “Direct Evidence for Chlorine-Enhanced Urban Ozone Formation in Houston, Texas.” Atmospheric Environment 37(9):1393–1400. https://doi.org/10.1016/S1352-2310(02)01007-5.
Theodoritsi, G. N., G. Ciarelli, and S. N. Pandis. 2021. “Simulation of the Evolution of Biomass Burning Organic Aerosol with Different Volatility Basis Set Schemes in PMCAMx-SRv1.0.” Geoscientific Model Development 14(4):2041–2055. https://doi.org/10.5194/gmd-14-2041-2021.
Theys, N., R. Volkamer, J. F. Müller, K. J. Zarzana, N. Kille, L. Clarisse, I. De Smedt, C. Lerot, H. Finkenzeller, F. Hendrick, T. K. Koenig, C. F. Lee, C. Knote, H. Yu, and M. Van Roozendael. 2020. “Global Nitrous Acid Emissions and Levels of Regional Oxidants Enhanced by Wildfires.” Nature Geoscience 13(10):681–686. https://doi.org/10.1038/s41561-020-0637-7.
Thornton, J. A., J. P. Kercher, T. P. Riedel, N. L. Wagner, J. Cozic, J. S. Holloway, W. P. Dubé, G. M. Wolfe, P. K. Quinn, A. M. Middlebrook, B. Alexander, and S. S. Brown. 2010. “A Large Atomic Chlorine Source Inferred from Mid-continental Reactive Nitrogen Chemistry.” Nature 464(7286):271–274. https://doi.org/10.1038/nature08905.
Tilgner, A., P. Bräuer, R. Wolke, and H. Herrmann. 2013. “Modelling Multiphase Chemistry in Deliquescent Aerosols and Clouds Using CAPRAM3.0i.” Journal of Atmospheric Chemistry 70(3):221–256. https://doi.org/10.1007/s10874-013-9267-4.
Tilgner, A., T. Schaefer, B. Alexander, M. Barth, J. L. Collett, Jr., K. M. Fahey, A. Nenes, H. O. T. Pye, H. Herrmann, and V. F. McNeill. 2021. “Acidity and the Multiphase Chemistry of Atmospheric Aqueous Particles and Clouds.” Atmospheric Chemistry and Physics 21(17):13483–13536. https://doi.org/10.5194/acp-21-13483-2021.
Tkacik, D. S., A. A. Presto, N. M. Donahue, and A. L. Robinson. 2012. “Secondary Organic Aerosol Formation from Intermediate-Volatility Organic Compounds: Cyclic, Linear, and Branched Alkanes.” Environmental Science & Technology 46(16):8773–8781. https://doi.org/10.1021/es301112c.
Tomaz, S., T. Cui, Y. Chen, K. G. Sexton, J. M. Roberts, C. Warneke, R. J. Yokelson, J. D. Surratt, and B. J. Turpin. 2018. “Photochemical Cloud Processing of Primary Wildfire Emissions as a Potential Source of Secondary Organic Aerosol.” Environmental Science & Technology 52(19):11027–11037. https://doi.org/10.1021/acs.est.8b03293.
Tuet, W. Y., Y. Chen, S. Fok, D. Gao, R. J. Weber, J. A. Champion, and N. L. Ng. 2017. “Chemical and Cellular Oxidant Production Induced by Naphthalene Secondary Organic Aerosol (SOA): Effect of Redox-Active Metals and Photochemical Aging.” Scientific Reports 7(1):15157. https://doi.org/10.1038/s41598-017-15071-8.
US Environmental Protection Agency. 2021. EPI Suite™-Estimation Program Interface. https://www.epa.gov/tsca-screening-tools/epi-suitetm-estimation-program-interface (accessed December 22, 2021).
Veres, P., J. M. Roberts, I. R. Burling, C. Warneke, J. de Gouw, and R. J. Yokelson. 2010. “Measurements of Gas-Phase Inorganic and Organic Acids from Biomass Fires by Negative-Ion Proton-Transfer Chemical-Ionization Mass Spectrometry.” Journal of Geophysical Research: Atmospheres 115(D23). https://doi.org/10.1029/2010JD014033.
Vidović, K., A. Kroflič, M. Šala, and I. Grgić. 2020. “Aqueous-Phase Brown Carbon Formation from Aromatic Precursors under Sunlight Conditions.” Atmosphere 11(2):131. https://doi.org/10.3390/atmos11020131.
Volkamer, R., P. Ziemann, and M. Molina. 2009. “Secondary Organic Aerosol Formation from Acetylene (C2H2): Seed Effect on SOA Yields Due to Organic Photochemistry in the Aerosol Aqueous Phase.” Atmospheric Chemistry and Physics 9(6):1907–1928. https://doi.org/10.5194/acp-9-1907-2009.
Wallington, T. J., M. D. Hurley, J. Xia, D. J. Wuebbles, S. Sillman, A. Ito, J. E. Penner, D. A. Ellis, J. Martin, S. A. Mabury, O. J. Nielsen, and M. P. Sulbaek Andersen. 2006. “Formation of C7F15COOH (PFOA) and Other Perfluorocarboxylic Acids during the Atmospheric Oxidation of 8:2 Fluorotelomer Alcohol.” Environmental Science & Technology 40(3):924–930. https://doi.org/10.1021/es051858x.
Wang, Z., J. C. DeWitt, C. P. Higgins, and I. T. Cousins. 2017. “A Never-Ending Story of Per- and Polyfluoroalkyl Substances (PFASs)?” Environmental Science & Technology 51(5):2508–2518.
Wei, Y., X. Chen, H. Chen, Y. Sun, W. Yang, H. Du, … and Z. Wang. 2021. “Investigating the Importance of Sub-grid Particle Formation in Point Source Plumes over Eastern China using IAP-AACM v1.0 with a Sub-grid Parameterization.” Geoscientific Model Development 14(7):4411–4428.
Whitten, G. Z., G. Heo, Y. Kimura, E. McDonald-Buller, D. T. Allen, W. P. L. Carter, and G. Yarwood. 2010. “A New Condensed Toluene Mechanism for Carbon Bond: CB05-TU.” Atmospheric Environment 44(40):5346–5355. https://doi.org/10.1016/j.atmosenv.2009.12.029.
Wilkins, J. L., G. Pouliot, K. Foley, W. Appel, and T. Pierce. 2018. “The Impact of US Wildland Fires on Ozone and Particulate Matter: A Comparison of Measurements and CMAQ Model Predictions from 2008 to 2012.” International Journal of Wildland Fire 27(10):684–698. https://doi.org/10.1071/wf18053.
Wong, J. P. S., M. Tsagkaraki, I. Tsiodra, N. Mihalopoulos, K. Violaki, M. Kanakidou, J. Sciare, A. Nenes, and R. J. Weber. 2019. “Effects of Atmospheric Processing on the Oxidative Potential of Biomass Burning Organic Aerosols.” Environmental Science & Technology 53(12):6747–6756. https://doi.org/10.1021/acs.est.9b01034.
Xu, L., J. D. Crounse, K. T. Vasquez, H. Allen, P. O. Wennberg, I. Bourgeois, … and R. J. Yokelson. 2021. “Ozone Chemistry in Western US Wildfire Plumes.” Science Advances 7(50):eabl3648.
Ye, X., P. Arab, and R. Ahmadov. 2021. “Evaluation and Intercomparison of Wildfire Smoke Forecasts from Multiple Modeling Systems for the 2019 Williams Flats Fire.” Atmospheric Chemistry and Physics 21(18):14427–14469. https://doi.org/10.5194/acp-21-14427-2021.
Yee, L., K. Kautzman, C. Loza, K. Schilling, M. Coggon, P. Chhabra, M. Chan, A. Chan, S. Hersey, and J. Crounse. 2013. “Secondary Organic Aerosol Formation from Biomass Burning Intermediates: Phenol and Methoxyphenols.” Atmospheric Chemistry and Physics 13(16):8019–8043. https://doi.org/10.5194/acp-13-8019-2013.
Ying, Q., J. Li, and S. H. Kota. 2015. “Significant Contributions of Isoprene to Summertime Secondary Organic Aerosol in Eastern United States.” Environmental Science & Technology 49(13):7834–7842. https://doi.org/10.1021/acs.est.5b02514.
Yokelson, R. J., I. T. Bertschi, T. J. Christian, P. V. Hobbs, D. E. Ward, and W. M. Hao. 2003. “Trace Gas Measurements in Nascent, Aged, and Cloud-Processed Smoke from African Savanna Fires by Airborne Fourier Transform Infrared Spectroscopy (AFTIR).” Journal of Geophysical Research: Atmospheres 108(D13). https://doi.org/10.1029/2002JD002322.
Yokelson, R. J., J. Crounse, P. DeCarlo, T. Karl, S. Urbanski, E. Atlas, T. Campos, Y. Shinozuka, V. Kapustin, and A. Clarke. 2009. “Emissions from Biomass Burning in the Yucatan.” Atmospheric Chemistry and Physics 9(15):5785–5812. https://doi.org/10.5194/acp-9-5785-2009.
Young, C. J., V. I. Furdui, J. Franklin, R. M. Koerner, D. C. Muir, and S. A. Mabury. 2007. “Perfluorinated Acids in Arctic Snow: New Evidence for Atmospheric Formation.” Environmental Science & Technology 41(10):3455–3461.
Young, C., R. Washenfelder, P. Edwards, D. Parrish, J. Gilman, W. Kuster, L. Mielke, H. Osthoff, C. Tsai, and O. Pikelnaya. 2014. “Chlorine as a Primary Radical: Evaluation of Methods to Understand Its Role in Initiation of Oxidative Cycles.” Atmospheric Chemistry and Physics 14(7):3427–3440. https://doi.org/10.5194/acp-14-3427-2014.
Yu, L., J. Smith, A. Laskin, C. Anastasio, J. Laskin, and Q. Zhang. 2014. “Chemical Characterization of SOA Formed from Aqueous-Phase Reactions of Phenols with the Triplet Excited State of Carbonyl and Hydroxyl Radical.” Atmospheric Chemistry and Physics 14(24):13801–13816. https://doi.org/10.5194/acp-14-13801-2014.
Yu, L., J. Smith, A. Laskin, K. M. George, C. Anastasio, J. Laskin, A. M. Dillner, and Q. Zhang. 2016. “Molecular Transformations of Phenolic SOA during Photochemical Aging in the Aqueous Phase: Competition among Oligomerization, Functionalization, and Fragmentation.” Atmospheric Chemistry and Physics 16(7):4511–4527. https://doi.org/10.5194/acp-16-4511-2016.
Yu, X., D. Li, D. Li, G. Zhang, H. Zhou, S. Li, W. Song, Y. Zhang, X. Bi, and J. Yu. 2020. “Enhanced Wet Deposition of Water-Soluble Organic Nitrogen During the Harvest Season: Influence of Biomass Burning and In-Cloud Scavenging.” Journal of Geophysical Research: Atmospheres 125(18):e2020JD032699. https://doi.org/10.1029/2020JD032699.
Zhang, Y., H. Forrister, J. Liu, J. Dibb, B. Anderson, J. P. Schwarz, A. E. Perring, J. L. Jimenez, P. Campuzano-Jost, and Y. Wang. 2017. “Top-of-Atmosphere Radiative Forcing Affected by Brown Carbon in the Upper Troposphere.” Nature Geoscience 10(7):486–489. https://doi.org/10.1038/ngeo2960.
Zhao, R., E. L. Mungall, A. K. Lee, D. Aljawhary, and J. P. Abbatt. 2014a. “Aqueous-Phase Photooxidation of Levoglucosan–A Mechanistic Study Using Aerosol Time-of-Flight Chemical Ionization Mass Spectrometry (Aerosol ToF-CIMS).” Atmospheric Chemistry and Physics 14(18):9695–9706. https://doi.org/10.5194/acp-14-9695-2014.
Zhao, Y., C. J. Hennigan, A. A. May, D. S. Tkacik, J. A. de Gouw, J. B. Gilman, W. C. Kuster, A. Borbon, and A. L. Robinson. 2014b. “Intermediate-Volatility Organic Compounds: A Large Source of Secondary Organic Aerosol.” Environmental Science & Technology 48(23):13743–13750. https://doi.org/10.1021/es5035188.
Zhou, S., S. Collier, D. A. Jaffe, N. L. Briggs, J. Hee, A. J. Sedlacek III, L. Kleinman, T. B. Onasch, and Q. Zhang. 2017. “Regional Influence of Wildfires on Aerosol Chemistry in the Western US and Insights into Atmospheric Aging of Biomass Burning Organic Aerosol.” Atmospheric Chemistry and Physics 17(3):2477–2493. https://doi.org/10.5194/acp-17-2477-2017.
Zuend, A., and J. H. Seinfeld. 2012. “Modeling the Gas-Particle Partitioning of Secondary Organic Aerosol: The Importance of Liquid-Liquid Phase Separation.” Atmospheric Chemistry and Physics 12(9):3857–3882. https://doi.org/10.5194/acp-12-3857-2012.
This page intentionally left blank.






























