
5
Myositis
Idiopathic inflammatory myopathies (IIMs), often collectively known as “myositis,” are rare diseases characterized by inflammation and weakness of the skeletal muscles which can also involve internal organs such as the lungs, heart, and esophagus. In clinical practice the most common myopathies are polymyositis (PM) and dermatomyositis (DM). In recent years, the classification of such diseases has broadened beyond PM and DM. The currently accepted diagnostic categories are PM, DM, necrotizing autoimmune myopathy (NAM, also known as immune-mediated necrotizing myopathy, or IMNM), antisynthetase syndrome, overlap myositis, inclusion body myositis, and juvenile myositis (Glaubitz et al., 2020; Lilleker and Chinoy, 2020). This chapter focuses on the more commonly known phenotypes: adult dermatomyositis, adult polymyositis, and juvenile dermatomyositis (JDM).
Overlap myositis (myositis that overlaps with other autoimmune diseases such as scleroderma, Sjögrens disease, and rheumatoid arthritis) likely accounts for the largest group of the myositis forms, with up to half of the cases, followed by DM with over one-third of the cases (Senécal et al., 2017). DM has an incidence of 1.4 patients per 100,000 persons in the United States, based on age- and gender-adjusted rates from 2003–2008 (Furst et al., 2012). Whereas most of the syndromes occur primarily in middle-aged patients, DM also occurs in children and adolescents (Glaubitz et al., 2020). The average age of diagnosis of DM is bimodal, with juvenile DM (JDM) most commonly diagnosed between 4 and 14 years of age and adult DM diagnosed between 40 and 60 years
of age (Aussy et al., 2017). DM affects both genders with a two-to-one female-to-male ratio. DM is more common in African Americans, though all racial and ethnic groups are affected (Findlay et al., 2015). PM, which is now understood to encompass the more recently described entity of IMNM, or NAM, occurs in 3.8 per 100,000 persons in the United States, based on 2003–2008 data (this estimate may include overlap syndromes) (Furst et al., 2012). Most cases of PM are diagnosed in adults between 31 and 60 years of age. PM, like DM, is more common in women and among African Americans. Precise epidemiological data are difficult to generate due to varying methods of case identification, small study sizes, geographical variances, and changes in diagnostic criteria and recording practices (Tran et al., 2013).
The chapter begins with an overview of clinical features, disease characteristics, and diagnosis. It then covers disease course and prognosis, treatment and management, and disease-specific functional limitations. Finally, a section on individuals with JDM addresses differences in the clinical features, prognosis, and treatment of childhood-onset DM compared with adult DM.
CLINICAL FEATURES, DIAGNOSIS, AND DISEASE COURSE
DM is a rare disease with multiple organ involvement which is clinically heterogeneous and can be difficult to diagnose. DM primarily affects the skin, muscles, joints, and lungs. Skin lesions in DM play an important role in the diagnosis and can present with or prior to muscle weakness (Dalakas, 2020). In more than half of patients, skin lesions appear before muscle involvement by months or years (Cassius et al., 2019). DM is also strongly associated with cancers.
PM is also a rare, immune-mediated disease with multiple organ involvement in which muscle weakness is a predominant feature; however, there is usually no skin involvement. Many patients referred for PM have another disease, most often inclusion-body myositis (IBM), NAM/IMNM, the anti-synthetase syndrome, or an inflammatory dystrophy. PM is historically considered to be a diagnosis of exclusion, defined as a “subacute proximal myopathy in adults who do not have rash, family history of neuromuscular disease, exposure to myotoxic drugs (d-penicillamine, zidovudine), involvement of facial and extraocular muscles, endocrinopathy, or the clinical phenotype of IBM” (Dalakas, 2020, p. 290). Over the last two decades, the field has been moving away from considering PM to be an exclusionary diagnosis and toward recognition of individual subtypes, such as NAM/IMNM and the antisynthetase syndrome, as separate disorders.
Clinical Features
Muscle Manifestations
Approximately 80 percent of patients with DM have myopathy, or disease that affects the muscle tissue. Those 20 percent with DM-consistent skin findings but without myopathy have what is called clinically amyopathic dermatomyositis (CADM) (DeWane et al., 2020). PM is by definition myopathic, meaning 100 percent of patients with PM experience a myopathy at some point during their illness. The classic muscle manifestation in DM is acute or subacute onset of symmetric, proximal muscle weakness. Patients with PM tend to present with symmetric weakness in a proximal distribution in the upper and lower extremities, though involvement of distal muscles also occurs to a lesser degree. The myopathy is typically painless. Muscle tenderness and myalgia are also reported (McGrath et al., 2018).
Skin Manifestations
A typical presentation of skin manifestations for an adult with DM might be a blue-purple rash around the eye with edema; red rash on face, knees, elbows, malleoli, neck, anterior chest (in V-sign), back and shoulders (in shawl sign); and knuckles with a violet rash (Gottron’s rash) that evolves into scaling discolorations (Yang et al., 2019). Also characteristic are dilated capillary loops at the base of the fingernails, irregular and thickened cuticles, and cracked palmar fingertips (known colloquially as “mechanic’s hands”) (Dalakas, 2020). Figure 5-1 depicts the common skin manifestations of DM.
The severity of skin manifestations does not necessarily reflect the disease course or the severity of the myositis or associated systemic involvement (DeWane et al., 2020).
Systemic Effects
DM and PM are associated with a number of potentially disabling complications across multiple body systems, including the pulmonary, cardiac, gastrointestinal (GI), endocrine, and vascular systems. Interstitial lung disease (ILD) is the most common complication of IIM and occurs in approximately 23 percent of Americans with DM or PM (Sun et al., 2020). ILD is usually characterized by a dry cough, shortness of breath, fine inspiratory crackles on lung examination, and a restrictive pattern on pulmonary function testing (McGrath et al., 2018). Myositis-related ILD follows two main clinical patterns, depending on the disease course: chronic interstitial lung disease and rapidly progressive (RP-ILD). RP-ILD is associated with a
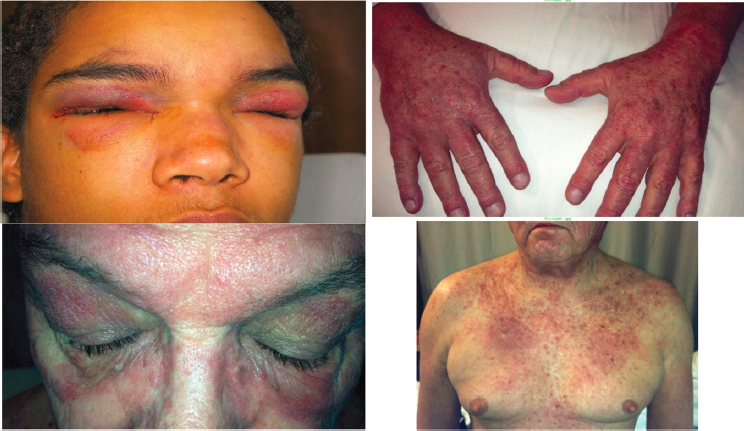
SOURCE: ACR, 2021, with permission.
poor prognosis, including rapid decline in lung function and mortality rates ranging from 70 to 90 percent (Barba et al., 2019). It is especially prevalent in Asian populations (DeWane et al., 2020). Anti-melanoma differentiation associated gene-5 (MDA-5) and anti-aminoacyl-tRNA synthetase (ARS) antibodies are the two myositis-specific antibodies most strongly associated with ILD (Mamyrova et al., 2021). ILD tends to occur less frequently in cases that are associated with cancer (McGrath et al., 2018). Other pulmonary complications include pulmonary hypertension (which may be secondary to severe or longstanding ILD) and serositis (inflammation of the tissue lining the heart, lungs, and peritoneum) (DeWane et al., 2020).
Cardiac manifestations such as conduction system abnormalities and heart failure are reported in as many as 30 percent of patients (McGrath et al., 2018). These can include conduction system abnormalities and arrhythmias, pericarditis, myocarditis, coronary artery disease, and congestive heart failure/diastolic dysfunction (DeWane et al., 2020; McGrath et al., 2018). Subclinical diastolic dysfunction is common in myositis patients. Cardiac involvement does not correlate with disease severity and may develop at any time (DeWane et al., 2020).
GI complications include dysphagia (difficulties with swallowing), aspiration of gastric contents, and delayed gastric emptying. Dysphagia is usually related to dysfunction of the pharynx or esophagus. Gastric and small intestinal motility may also be affected. In rare cases, vasculopathy may
lead to GI tract infarction or perforation (DeWane et al., 2020; McGrath et al., 2018).
Vascular complications may also occur. Particularly, cutaneous vasculopathy may cause ulcerations, especially in patients with antiMDA5 antibodies (DeWane et al., 2020).
Rheumatologic complications may include arthralgias, arthritis, and joint contractures (McGrath et al., 2018).
Cancers
The risk of malignancy (cancer) is increased in adult patients with DM to approximately 10 to 30 percent within 2 to 5 years of disease onset. Most cases occur in individuals over the age of 40 years (Dalakas, 2020; McGrath et al., 2018). An annual cancer screening is necessary in the first 3 years. The most common cancers associated with adult-onset DM include cancers of the blood and lymphatic systems, followed by cancers of the lung, colon, bladder, breast, ovary, cervix, pancreas, and esophagus (McGrath et al., 2018). Nasopharynx cancer is also associated with DM in Asian populations (Dalakas, 2020). Successfully treating the underlying malignancy can result in improved muscle strength (McGrath et al., 2018). Qiang and colleagues (2017) performed a systematic review and meta-analysis of malignancy risk in DM and PM which included five studies with a total of 4,538 DM or PM patients. They found an overall malignancy rate of 12.1 percent. By myositis diagnosis type, the prevalence of malignant disease that occurred concurrently with or after diagnosis was 14.8 percent for DM and 9.9 percent for PM. The overall relative risk for malignant disease was increased in both DM and PM, with a higher risk found in DM than in PM (Oldroyd et al., 2021).
Clinical Diagnosis and Professionally Accepted Classification Criteria
Standardized clinical practice guidelines do not currently exist for IIMs. A professionally accepted diagnosis is based on a combination of a patient’s history, clinical examination, and laboratory tests such as creatine kinase and muscle biopsy (Meyer et al., 2019). Additional testing can include myopathic features on electromyography and the detection of myositis autoantibodies; these modalities can confirm a clinically suspected diagnosis and help clinicians evaluate the risk of systemic involvement and myositis-associated cancers. The approach to additional testing for myopathy depends on diagnostic certainty. In patients with the characteristic skin findings of DM and proximal muscle weakness, additional testing may not be necessary. For others in whom there is more uncertainty, additional testing is needed to confirm the presence of myopathy. An initial
workup for DM often involves several different subspecialists as well as referral to a dermatologist, rheumatologist, pulmonologist, or neurologist, depending on the symptoms and clinical findings. The absence of skin involvement is a first indicator differentiating PM from DM. Muscle biopsy can also help distinguish PM from DM. One distinguishing histological feature in PM is that the cellular infiltrate in muscle is predominantly within the fascicle, in which there are increased numbers of cytotoxic CD8+ T cells (while in DM the cellular infiltrate is predominantly perifascicular and often perivascular) (Amato and Barohn, 2009). In both DM and PM, patients should be assessed for ILD and cancer.
In terms of classification criteria, the Bohan and Peter criteria (1975) are the most enduring criteria; however, in the past 10 years researchers have gradually shifted toward using revised criteria, given the advancements in the scientific understanding of myositis. The revised criteria authored by the European Alliance of Associations for Rheumatology (EULAR) and American College of Rheumatology (ACR) (Lundberg et al., 2017) provide specific classifications for inclusion body myositis and amyopathic dermatomyositis as well as distinctions between adult and childhood-onset disease. The EULAR/ACR criteria are now accepted as the standard in clinical trials. They are primarily used in clinical research studies and not used as a formal part of clinical practice diagnostic criteria, given that it may be cumbersome for practicing clinicians to complete the entire dataset and that the criteria have low sensitivity (87 percent as reported by Waldman et al., 2020) for diagnosing DM.
This section provides an overview of tools used in the clinical setting to diagnose DM and PM.
History and Physical Examination
Patients should be asked to describe their muscle weakness, including duration, severity, distribution, mode of onset, and location as well as their ability to carry out activities that they commonly perform, such as climbing stairs and carrying heavy groceries. Additionally, patients should describe any history of dysphagia, which may suggest esophageal involvement, and of cough or shortness of breath, which may occur due to pulmonary involvement. Because overlap syndrome can exist, patients should also be asked about symptoms of other systemic rheumatic diseases, particularly systemic lupus erythematosus and systemic sclerosis. Patients should be queried about whether they have had any symptoms suggestive of cancer (such as unexplained weight loss, abdominal pain or bloating, or persistent cough) and if they have experienced cutaneous eruptions, photosensitivity, pruritus, or Raynaud’s phenomenon. Finally, the use and timing of drugs that may cause myopathy, particularly statins, should be determined (Miller, 2021).
A standard physical examination primarily involves the skin, muscles, and joints. A skin examination should pay particular attention to the scalp, face, eyelids, hands, fingers, extensor surfaces, upper chest and back, lateral thighs, and joints. A detailed neurologic and neuromuscular examination is critical to determine the severity and distribution of weakness and of muscle tenderness as well as to uncover any other abnormal neurologic findings. A thorough joint examination should be performed to detect signs of inflammatory arthritis. A general physical examination should also be performed to detect systemic effects, with particular attention paid to the heart and lungs, for evidence of ILD (Miller, 2021).
Muscle Enzymes
Creatine kinase (CK) is a routinely used biomarker for myositis diagnosis and disease activity follow-up. Elevated serum levels of CK are the most sensitive indicator of muscle fiber damage in IIM, though an increase in CK can also be caused by physical exercise, drug use, and myocardial infarction (Cassius et al., 2019). CK levels are most elevated in necrotizing autoimmune myositis and least elevated in inclusion body myositis. Normal CK levels do not rule out a myositis diagnosis, as CK appears to be normal in some instances of myositis (Cassius et al., 2019; Sizemore, 2015). In cases of normal CK levels, it is helpful to investigate aldolase levels (Sizemore, 2015). Myositis should be suspected in patients with cutaneous eruption suggestive of myositis in the setting of elevated CK. However, CK level cannot be used on its own to diagnose myositis and must be interpreted in the context of the patient’s complete clinical workup, including myopathic pattern on electromyography and muscle and skin biopsy.
Electrophysiological Features
Electromyography (EMG), an electrodiagnostic medicine technique for evaluating and recording the electrical activity produced by skeletal muscles, plays an important role in diagnosing and managing myopathy. EMG studies are useful early in the disease and show abnormal findings in 70–90 percent of cases but are nonspecific to myositis, as an abnormal finding can indicate other muscle diseases (Marvi et al., 2012). EMG shows evidence of muscle membrane irritability, including increased insertional activity, spontaneous fibrillations, positive sharp waves, and complex repetitive discharges (McGrath et al., 2018). It is most helpful in distinguishing myopathic causes of weakness from neuropathic disorders and myasthenia gravis.
Imaging
Magnetic resonance imaging (MRI) is a sensitive technique for evaluating myositis with the presence of muscle inflammation, edema with active myositis, fibrosis, and calcification. In late disease it can show muscle atrophy (Marvi et al., 2012). Unlike biopsy, MRI can assess large areas of muscle, thereby avoiding issues of sampling error. Like EMG, it is nonspecific and may not distinguish inflammatory myopathy from other diseases such as muscular dystrophy and metabolic myopathy. If DM or PM is suggested by EMG or MRI, a muscle or skin biopsy is needed to make a definite diagnosis.
Muscle and Skin Biopsy
Muscle biopsy should be considered in the diagnosis of all adult patients with inflammatory myopathy. The histologic features of both DM and PM include muscle fiber necrosis, degeneration, regeneration, and an inflammatory cell infiltrate. DM can be distinguished from PM using muscle biopsy. In DM, cellular infiltrate is predominantly perifascicular and often perivascular. Other indicators of DM are B lymphocytes and an increased number of plasmacytoid dendritic cells (Marvi et al., 2012). In patients with typical DM presentation, a skin biopsy or test confirming myositis-specific autoantibodies may be sufficient to confirm a diagnosis of DM.
Choosing the optimal muscle to biopsy is based on the clinical examination. The selected muscle should be moderately weak but not severely weak because there is a limited diagnostic yield from biopsies of very weak muscles, which tend to show nonspecific end-stage fibro-fatty changes. EMG or MRI can help identify an appropriate muscle biopsy site in cases where patients are weak in muscles that are less routinely biopsied (McGrath et al., 2018).
Myositis Autoantibodies
Autoantibodies are proteins that target another one of a person’s own proteins; they appear in the blood when a person has an autoimmune disease. They are a useful emerging biomarker, as there is usually a close association between particular autoantibodies and particular subtypes of a disease or clinical symptoms that have a similar prognosis and treatment. Autoantibodies found in patients with myositis are classified into myositis-specific antibodies (MSAs) and myositis-associated antibodies (MAAs). MSAs are detected in between 30 percent and 50 percent of patients with DM (Cassius et al., 2019), although in some larger specialty centers the percentage of DM patients with autoantibodies can be as high as 80 percent.
DM-specific autoantibodies are rarely found in other diseases, and usually a patient only has one specific MSA, so the presence of a DM-specific autoantibody can confirm a diagnosis of DM in an adult patient (Cassius et al., 2019). Identifying particular MSAs can help clinicians predict disease course, for example by providing information about the risk of systemic effects and cancer.
Table 5-1 presents five classic autoantibodies that are currently recognized as being specific to DM. Anti-Mi-2 antibodies are found in 7–30 percent of patients with DM and are associated with acute onset of classic DM, particularly severe skin manifestations at initial presentation. Although DM associated with anti-Mi2 antibodies is thought to have a good response to therapy, a favorable prognosis, and a reduced risk of cancer compared with other patients with DM (McGrath et al., 2018), recent work has also shown that patients with anti-Mi2-positive DM have more severe muscle disease than patients with anti-Mi2-negative DM or patients with anti-synthetase syndrome (Pinal-Fernandez et al., 2019). Additionally, anti-Mi2 autoantibody levels correlate with disease severity and may normalize in patients who enter remission. Antibodies directed against melanoma differentiation association protein-5 (MDA-5) occur in approximately 15 percent of patients with DM. They are usually associated with minimal muscle involvement (amyopathic DM), skin ulcers, mechanic’s hand, and rapidly progressive ILD, particularly in Asian populations (Cassius et al., 2019; McGrath et al., 2018). Antibodies directed against transcriptional
TABLE 5-1 Dermatomyositis-Specific Myositis Autoantibodies
| Target of Antibodies | Prevalence in DM | Associated Clinical Presentation |
|---|---|---|
| Mi2 | 7–30% | Classical skin rash, reduced ILD and cancer risk, more severe muscle disease |
| MDA5 | ~15% | Skin ulcers, palmar papules, mechanic’s hands, minimal muscle involvement, higher risk of ILD |
| TIF1 | 14–31% | Classic skin eruption, poikiloderma, reduced ILD risk, increased cancer risk |
| NXP2 | 1.6–30% | Severe skin eruption, calcinosis, reduced ILD risk, severe muscle weakness and increased risk of cancer, generally responsive to treatment |
| SAE | 1–8% | Initial presentation with rash and amyopathic, later muscle involvement, varying systemic effects |
NOTE: ILD = interstitial lung disease; MDA = melanoma differentiation-associated gene; NXP = nuclear matrix protein; SAE = small ubiquitin-like modifier-activating enzyme; TIF = transcription intermediary factor.
SOURCES: Albayda et al., 2021; Cassius et al., 2019; McGrath et al., 2018.
intermediary factor 1-ϒ (TIF1-ϒ), also known as anti-p155/140 antibody, have a prevalence of 14 to 31 percent in DM and strongly predict the risk of cancer, showing 89 percent specificity and 78 percent sensitivity for cancer diagnosis, with a positive predictive value of 58 percent and a negative predictive value of 95 percent. In addition, these antibodies are associated with particularly severe skin manifestations, including palmar hyperkeratotic papules, psoriatic type lesions, and hypopigmented and telangiectatic skin patches (McGrath et al., 2018). Antibodies against nuclear matrix protein-2 (NXP-2), previously known as anti-MJ, are found in up to 30 percent of patients with DM and are associated with a younger age of onset, severe muscle weakness, calcinosis, good response to treatment, and an increased risk of cancer (Cassius et al., 2019; McGrath et al., 2018). Finally, antibodies directed against small ubiquitin-like modifier-activating enzyme (SAE) are reported to occur with a prevalence ranging from less than 1 to 8 percent of DM patients. SAE autoantibodies associate with a clinical phenotype of DM that most commonly presents with an initial rash, followed by muscle involvement and varying extramuscular involvement. Coincident cancer can be seen in anti-SAE-positive DM; thus, prudent malignancy screening may be warranted (Albayda et al., 2021).
Although this report focuses on DM and PM, it is also worth noting that the antisynthetase syndrome is increasingly recognized as myositis autoantibodies are more frequently ordered. In particular, Jo-1 antibodies are strongly associated with the following conditions that define the phenotype known as the antisynthetase syndrome: ILD (66 percent of patients), arthralgias (56 percent), fever (27 percent), Raynaud’s phenomenon (40 percent), and mechanic’s hands (31 percent) (McGrath et al., 2018). While these patients may also have a rash similar to DM, the antisynthetase syndrome is now known to exist as its own myositis subtype separate from DM and PM. Additionally, recent research suggests that several non–Jo-1 ARS autoantibodies may be present in patients with ILD with subclinical myositis.
There is much research and standardization still to be done in the field of MSAs. MSAs are useful as additional evidence to confirm a diagnosis and to help clinicians select treatments, predict treatment response, and predict disease course. They should, however, not be used as the only tool for the diagnosis of myositis and must be interpreted in the context of the clinical features present.
Disease Course, Outcomes, and Variability
The usual course of myositis is progressive, but it can be relapsing and remitting with periods of worsening and quiescence. The disease course can be categorized as monophasic, meaning a disease in which the patient can eventually be tapered off medication; chronic, meaning
the patient continues to require medication; or polyphasic, meaning that the patient experiences relapse and remission. The typical trajectory of myositis begins with weakness over days to weeks, or it can present more slowly over a few months. The proximal arms and legs are usually affected. Neck flexors are also often involved early. In up to 30 percent of individuals, weakness of oropharyngeal and esophageal muscles results in dysphagia. Dysarthria and facial muscle weakness can occur but are uncommon (McGrath et al., 2018). Dysphagia, dysphonia, and aspiration are associated with poor prognosis (Ogawa-Momohara et al., 2019). The characteristic skin rash can develop months after disease onset, but more typically it accompanies or precedes the onset of muscle weakness (McGrath et al., 2018). In DM, skin manifestations vary, and their clinical course may or may not parallel that of muscle disease and systemic involvement in time course or severity (DeWane et al., 2020).
The disease course and outcomes vary, and prognosis depends on systemic effects and multimorbidity. Most patients respond to immunosuppressive therapies, but their symptoms may not resolve completely (McGrath et al., 2018). The disease may go into remission, but often patients must remain on longstanding immunosuppressive therapies (McGrath et al., 2018). In cases where muscle inflammation leads to irreversible muscle atrophy and fatty replacement, permanent muscle weakness in those targeted muscle groups will persist due to the permanent damage. ILD and RP-ILD are well-recognized complications of DM and PM and worsen the prognosis of the disease. There is substantial variation in the progression of disease among patients with IIM-associated ILD (McGrath et al., 2018). Primary cardiac involvement in IIM and particularly DM is common and often subclinical and associated with poor prognosis. (Cassius et al., 2019). Other potential longstanding complications include joint deformities and contractures, which may occur more often in children. Other accompanying autoimmune features such as Sjögrens syndrome–associated sicca symptoms may remain (Glaubitz et al., 2020). Despite control of the disease, many patients are left with damage such as calcinosis, muscle atrophy, and fatty replacement, which can lead to permanent weakness, and decreased endurance as measured by the functional index 2 (FI2) test (Rider et al., 2011). Even patients with seemingly normal muscle strength can have considerable reduction in endurance noted on FI2 testing (Amici et al., 2019).
Patients with myositis can have episodic flares that can be unpredictable, and the quality of life measures may be reduced for them, leading to both depression and anxiety. The unpredictable nature of flares can lead patients to have difficulty with sustained employment and can affect interpersonal relationships. In severe cases where damage ensues, patients are dependent on assistive devices such as canes or wheelchairs, and patients can become increasingly dependent on their partners as caregivers.
Furthermore, treatments may lead to opportunistic infections or cancer (Redondo-Benito et al., 2018).
In the clinical setting, evaluations of disease activity and severity are based on the degree of potentially reversible inflammation that might respond to immunosuppressive treatments (disease activity) and the degree of fibrosis or scarring resulting in nonreversible tissue injury and damage that will not respond to treatment (Espinosa-Ortega et al., 2017). Validated myositis-specific disease flare and assessment tools do not currently exist for the clinical setting, so clinicians mostly use cutaneous-, arthritis- or neurology-specific measures to assess severity in patients. A few myositis-specific outcome measures have been developed and validated recently for use in research settings (Espinosa-Ortega et al., 2017).
There is a validated ACR/EULAR Criteria for Minimal, Moderate, and Major Clinical Response developed by the International Myositis Assessment and Clinical Studies Group (IMACS) which is intended for use in myositis clinical trials (Lundberg et al., 2017). The IMACS criteria employ six core set measures. Summing the scores in those six core set measures results in a total improvement score which can be used to gauge improvement or worsening of disease over time (Aggarwal et al., 2017). The thresholds differ for adults and children. For each population, a total improvement score on a scale of 0–100 is intended to provide a quantitative assessment of the degree of response for each patient. This score is the sum of improvement in each of the following six core set measures: physician global activity, patient global activity, manual muscle testing, the Health Assessment Questionnaire, enzymes, and extramuscular activity in adults (Aggarwal et al., 2017)
The “extra-muscular disease activity” component of the core set represents a prominent aspect of disease activity in myositis, as systemic effects can have a greater effect on disease severity than does muscular inflammation. Lung computed tomography, pulmonary functional tests, echocardiography, heart MRI, and esophageal motility assessment are useful tools in assessing extra-muscular disease activity and severity (Espinosa-Ortega et al., 2017).
In addition, a myositis-specific patient-reported outcome tool is presently being developed by the OMERACT Myositis working group (Aggarwal et al., 2017; Alexanderson et al., 2014). The tool will include health-related quality-of-life measures and reflect patient perspectives. Imaging and immunologic biomarkers provide objective measures that discriminate disease activity and damage, but they need to be validated in clinical trials (Rider et al., 2018).
TREATMENT AND MANAGEMENT
Treatment strategies are similar for DM and PM. The goal of treatment is to improve muscle strength, ameliorate organ manifestations, and reduce
pain (Glaubitz et al., 2020). Currently, treatments are prescribed according to expert consensus, as evidence-based guidelines for treatments do not exist (Aggarwal et al., 2017; Barsotti and Lundberg, 2018; Griger et al., 2017). Owing to the rarity of the disease and the limitations of large double-blind placebo controlled trails, few agents are FDA-approved for myositis. To date, only corticosteroids, Acthar, and most recently IVIG Octagam 10 percent are currently approved (Aggarwal et al., 2021; Barsotti and Lundberg, 2018). Unfortunately, practitioners often find it difficult to prescribe therapies because of the off-label nature of these therapeutic agents, which are rarely included in an insurance formulary or the Medicare Compendium. With limited FDA-approved therapies, it is challenging for patients to obtain coverage, and some patients who demonstrate efficacy are forced to stop their medications because of a new insurance rule or change in insurance or Centers for Medicare & Medicaid Services rules. Despite this and the lack of placebo-controlled trials, glucocorticoids are considered to be the mainstay of initial management (Griger et al., 2017). Early recognition and intervention are essential to ameliorating disease outcome. Often, immunosuppressive therapies, especially steroid-sparing agents may take weeks to months to take effect and may need to be used in combination.
Several longstanding agents are used in a stepwise fashion in routine practice. Corticosteroids with adjunctive steroid-sparing immunosuppressive therapies are recommended to treat disease activity, prevent mortality, and reduce long-term disability. The basic treatment includes a combination of glucocorticoids with another immunosuppressive agent to control refractory disease, disease flares, and skin involvement and to reduce the risk of glucocorticoid (GC)-related side effects (Barsotti and Lundberg, 2018). Conventional immunosuppressive drugs (known as disease-modifying anti-rheumatic drugs, or DMARDs) that are used in first-line therapy include methotrexate, azathioprine, and mycophenolate mofetil (Glaubitz et al., 2020). They have a steroid-sparing effect and can treat extramuscular manifestations (Griger et al., 2017). Methotrexate has also been successfully used in the treatment of the skin manifestations of DM, although with conflicting results (Barsotti and Lundberg, 2018). A randomized controlled trial (RCT) conducted in 1980 concluded that azathioprine in combination with GCs was associated with better functional outcome in patients with PM (Bunch, 1981; Bunch et al., 1980); combination therapy continues to be recommended (Oddis and Aggarwal, 2018). Intravenous immunoglobulin (IVIG) can be considered as an alternative option or additional treatment in cases where corticosteroids and standard immunosuppressants are not sufficiently effective or are contraindicated. IVIG has been reported to be effective in treating refractory ILD, skin manifestations, and esophageal involvement in addition to treating muscular symptoms (Glaubitz et al., 2020).
Second-line therapy includes cyclosporine-A or tacrolimus. Therapy escalation—for example, in response to persistent weakness or organ involvement—can involve additional treatment with rituximab, cyclophosphamide, or mycophenolate mofetil, especially in cases with ILD (Glaubitz et al., 2020). Rituximab is the most commonly used biologic in the treatment of IIM (Barsotti and Lundberg, 2018). The main indications for rituximab treatment include refractory muscular, lung, skin, or joint involvement. Patients with myositis-specific autoantibodies seem to have a better response to the treatment, in particular those with anti-ARS positivity, anti-signal recognition particle, and Mi-2 antibodies. Rituximab has been shown to reduce disease activity and to have a steroid-sparing effect (Barsotti and Lundberg, 2018).
The addition of experimental third-line therapies, such as leflunomide, tofacitinib, and baricitinib, is indicated in severe, refractory, or corticosteroid-dependent diseases (Glaubitz et al., 2020; Griger et al., 2017).
In general, immunosuppressants should be chosen according to the patient’s clinical features and systemic involvement (see Table 5-2).
Future trials may consider subgrouping patients into clinical and serological subtypes to help identify biomarkers for response to specific immunosuppressive and biological agents (Barsotti and Lundberg, 2018).
Likelihood of Improvement with Treatment
Prompt diagnosis and treatment generally leads to better outcomes, decreased permanent damage, and decreased long-term disability. GC treatment is able to reduce muscular inflammation, and more than 60 percent of the patients show improvement of muscle symptoms when treated with GCs. This occurs in particular in the first 6 months after the start of the
TABLE 5-2 Common Drugs Indicated for the Treatment of Myositis and Their Efficacy According to Clinical Features and Systemic Involvement
| Clinical Features | Treatment | ||||||
|---|---|---|---|---|---|---|---|
| GCs | Rituximab | Methotrexate | Azathioprine | HCQ | IVIG | MMF | |
| Muscular | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Skin | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |
| ILD | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |
| Arthritis | ✓ | ✓ | ✓ | ✓ | |||
| Dysphagia | ✓ | ✓ | ✓ | ✓ | |||
NOTE: GCs = glucocorticoids; HCQ = hydroxychloroquine; IVIG = intravenous immunoglobulin (antibodies administered intravenously); MMF = mycophenolate mofetil.
SOURCES: Aggarwal et al., 2017; Barsotti and Lundberg, 2018; Griger et al., 2017.
treatment. High GC doses have also been used successfully for lung involvement (Barsotti and Lundberg, 2018).
Gordon and colleagues (2012) conducted a systematic review of RCTs or quasi-RCTs to assess the effects of immunosuppressant or immunomodulatory treatments on outcomes in patients with DM and PM. The two primary outcomes were change in function or a disability scale measured as the proportion of participants who improved by one or two grades, defined using IMACS criteria for improvement and time to relapse or remission, or based on a 15 percent or greater improvement in muscle strength after 6 months compared with baseline. The systematic review highlighted the lack of high-quality RCTs that answered this question and concluded that there was insufficient evidence from available RCTs to confirm whether the immunosuppressant treatments tested lead to outcomes improvement. The authors noted that conducting high-quality RCTs in rare diseases is difficult since they require sufficient sample sizes to deliver statistical power and individual clinicians rarely have the sufficient number of participants to conduct trials. These diseases may take months to years to respond fully and may require several therapeutic interventions before finding the optimal treatment strategy for each individual patient.
Secondary Impairments from Treatment
Secondary impairments from therapies are described in Chapter 2. Specifically in the case of myositis, longstanding GC therapy can lead to diabetes, cataracts, glaucoma, skin fragility, osteoporosis, and even steroid myopathy, which can further compound the difficulty in treating a patient with underlying myopathy (Barsotti and Lundberg, 2018).
Select Treatments Currently in Clinical Trials
There are clinical trials in progress for therapeutic interventions for myositis. A search on the National Institutes of Health’s (NIH’s) National Library of Medicine website on October 22, 2021, found 107 studies for myositis in the United States. The committee used the following filters in the search: all studies, United States only, all ages. Several novel approaches to treating DM have recently garnered significant interest. Novel therapeutic approaches targeting specific immunological pathways are highly promising. In order to evaluate their efficacy, large RCTs are needed (Glaubitz et al., 2020). For example, there is robust evidence that janus kinase inhibitors (JKIs) are effective in treating cutaneous DM, which is driven by type I interferons. Both in vivo and in vitro data have shown that JKIs decrease the levels of type I interferons in individuals with DM. Case series have shown that several JKIs are effective for treating refractory cutaneous
disease. One series has also shown that JKIs may be an effective add-on therapy in patients with RP-ILD. A recent open-label clinical trial found tofacitinib to be effective in treating refractory DM, notably in patients with skin disease (Paik et al., 2021). Further research on the pathogenesis of myositis is essential to understand the different phenotypes and to better predict the response to a specific treatment. Table 5-3 describes selected agents that have been previously studied or are currently being studied in clinical trials.
DISEASE-SPECIFIC FUNCTIONAL LIMITATIONS
Functional disability is common in individuals with myositis (Espinosa-Ortega et al., 2017; Rider et al., 2018). Of the inflammatory myopathies, the most prominent clinical feature is impaired muscle function, predominantly in the proximal and axial muscle groups, that results in functional limitations related to muscle weakness and decreased endurance with more fatigability. Muscle weakness can be assessed by standardized motor tasks or estimated by questionnaires and scales (Rider et al., 2018). The manual muscle test (MMT) measures isometric muscle strength as part of a physical examination (Rider et al., 2018). The Myositis Functional Index-2 is another muscle endurance test that can be used to identify muscle impairment, and it provides more accurate results than MMT in patients with high MMT scores (Amici et al., 2019).
Functional limitation in myositis can be measured, for example, using the six core set measures described by IMACS, which are related to disease activity, damage, and measures of patient-reported outcomes (Aggarwal et al., 2017; Rider et al., 2018). However, these measures are generally not used in a clinical setting. From a patient perspective, fatigue, pain, and physical function impairments also play a large role in disability, a theme that has been identified recently by the OMERACT myositis working group (Alexanderson et al., 2014; Espinosa-Ortega et al., 2017).
JUVENILE DERMATOMYOSITIS
Juvenile myositis shares many of the clinical features of the adult forms of the disease with similar immunopathogenesis, although there are some important differences. All of the juvenile idiopathic inflammatory myopathies (JIIMs) are rare. The most common form is JDM, which accounts for approximately 80 percent of all juvenile inflammatory myopathies (Rider and Nistala, 2016), for a prevalence of approximately 2.5 per 100,000 and an incidence of approximately 2.7 per million children per year between 1994–2003 (Meyer et al., 2015). Juvenile polymyositis (JPM) is extremely rare and often mimics disease in adults. It is estimated to be
TABLE 5-3 PM/DM Clinical Trials Overview
| Drug | Mechanism of Action | Clinical Trial Registration | Notes |
|---|---|---|---|
| Completed, published | |||
| Rituximab | Anti-CD20 | NCT00106184 | Although the primary endpoint was not reached, a majority of patients (83%) showed clinical improvement and steroid-sparing effect during the trial |
| Sifalimumab | Anti-interferon alpha | NCT00533091 | Suppression of the IFN signature in blood and muscle tissue in myositis patients, which correlated with clinical improvement |
| ACTH Gel, RCI | Adrenocorticotropic hormone | NCT01906372 | Reduced steroid dosing after 24 weeks |
| IVIG | Immunomodulator | NCT00001261 | |
| Etanercept | TNF alpha Inhibitors | NCT00112385 | |
| Infliximab | NCT00033891 | ||
| Abatacept | T-cell activation inhibitor | NCT01315938 | |
| Tofacitinib | JAK 1/3 inhibitor | NCT03002649 | |
| Octagam | IVIG/immunomodulator | NCT0272875 | |
| Completed, unpublished | |||
| Abatacept | T-cell activation inhibitor | NCT02971683 | |
| Eculizumab | C5 inhibitor | NCT00005571 | |
| JBT-101(Anabasum) | Endocanabinoid receptor agonist | NCT02466243 | |
| Toclizumab | IL-6 antagonist | NCT02043548 | |
| Lenabasum | Endocanabinoid receptor agonist | NCT03813160 | |
| Drug | Mechanism of Action | Clinical Trial Registration | Notes |
|---|---|---|---|
| In Process | |||
| PF-06823859 | Humanized Ig neutralizing Ab against INFB | NCT03181893 | |
| KZR-616 | Immunoproteasome inhibitor | NCT04033926 | |
| Belimumab | B-cell activation inhibitor | NCT02347891 | |
| Hizentra | SQ immunoglobulin/immunomodulator | NCT02271165 | |
| Alpremilast | PDE-4 inhibitor | NCT03529955 | |
SOURCES: Aggarwal et al., 2018; Dalakas et al., 1993; Liao et al., 2011; Muscle Study Group, 2011; Oddis et al., 2013; Paik et al., 2021; Tjärnlund et al., 2018.
about one-tenth as common as JDM (Meyer et al., 2015). This section will focus on JDM and highlights differences that exist in diagnosis and treatment between JDM and adult DM.
Clinical Features, Classification, and Disease Course
Clinical Features
JDM manifests with a typical skin rash as the hallmark feature, along with symmetric proximal muscle weakness (Huber, 2018). The average age of onset in the United States is 7 years, with 25 percent of patients presenting younger than 4 years of age at disease onset. The ratio of girls to boys is 2.3 to 1 (Feldman et al., 2008).
The disease manifests as a spectrum, with some children having more significant muscle inflammation, some with equal skin and muscle disease, and others predominantly with skin inflammation. While skin findings are a requirement to make the diagnosis, a small subset of children have undetectable muscle involvement (Mamyrova et al., 2018) and are referred to as amyopathic, hypomyopathic, skin predominant, or juvenile dermatomyositis sine myositis. Approximately 25 percent of those children will go on to develop more typical JDM over time (Gerami et al., 2007).
Systemic manifestations include ILD (6 percent of cases), cardiac involvement (uncommon), GI symptoms such as dysphagia and dysmotility (5–37 percent of cases), lipodystrophy (10–30 percent of patients), vasculitis, calcinosis, and arthritis (DeWane et al., 2020; Robinson et al., 2014). Notably, compared with adults with DM, in JDM patients ILD and cancer are more rare, while GI, endocrine, and vascular effects are more common (DeWane et al., 2020). Calcinosis is more common in JDM than in adult DM, and occurs in approximately one-third of JDM patients (Hoeltzel et al., 2014). Calcinosis can cause long-term complications, including severe joint contractures and immobility. It can be considered a marker of disease severity and possibly inadequate treatment, as it has the potential to be prevented through early, aggressive immunotherapy (Hoeltzel et al., 2014).
Diagnosis and Classification
Myositis will be suspected when patients have muscle weakness upon physical examination or elevated muscle enzymes, and cross-sectional imaging with an MRI may not be performed (Huber, 2018). An experienced pediatric rheumatologist will typically make the diagnosis of JDM after confirming myositis (by some combination of physical exam, documenting muscle enzyme elevation, and MRI) in a child with typical skin rashes and
proximal muscle weakness. EMG and a muscle biopsy may be performed when there is diagnostic uncertainty (e.g., to look for a neuromuscular cause) or when another JIIM aside from JDM is suspected, or it may still be the standard diagnostic approach of the treating physician. In contrast, for children with JPM, muscle imaging with an MRI and muscle biopsy are almost always performed.
Approximately 50 percent of children undergo a muscle biopsy, and only 30 percent undergo EMG (Robinson et al., 2014). Because of its widespread availability, MRI has replaced those other modalities for documenting myositis. MRI is used diagnostically in 90 percent of cases of JDM. JDM is almost never associated with malignancy, as is the case in adults, and therefore screening for malignancy is not standard practice (Morris and Dare, 2010). That may also explain why muscle biopsy is not always performed, as malignancy typically does not need to be excluded.
When a muscle biopsy is performed, there is considerable histologic overlap between DM in adults and children, but perifascicular atrophy seen on a biopsy specimen of muscle may be more reliably identified in JDM (DeWane et al., 2020). In addition, vascular involvement in JDM is often more prominent (Pestronk, 2011).
It is important to note that myositis can occur as a feature of a larger systemic autoimmune disease, such as systemic lupus erythematosus, where myositis is just one of many disease manifestations.
Most studies report delays in diagnosis, averaging approximately 6 months from disease onset to diagnosis. However, much longer delays are common. It is important to make the diagnosis of JIIM in a timely fashion because delays in diagnosis are associated with important negative outcomes, including increased risks of calcinosis and mortality (Huber, 2018).
Disease Course and Outcomes
The disease course for JDM is variable. Historically, JDM led to significant morbidity and mortality, and approximately one-third of patients have died due to the disease and another one-third developed permanent severe disability (Ramanan and Feldman, 2002). Since the advent of GCs and modern immune modulation, however, the prognosis has changed considerably for many patients; though disease morbidity remains high, mortality is now a rare outcome.
There are only a few studies on long-term outcomes in children with JDM. A Canadian cohort of 65 patients found that 37 percent were off medication by 24 months (monophasic), half had a chronic course requiring medication at 24 months, and 11 percent were having relapses and remission (polyphasic) (Huber et al., 2001). A large cohort of 490 children
from centers in Italy, Brazil, Argentina, Mexico, and the United Kingdom reported 41 percent being off medication by 24 months and the rest having a chronic course requiring medication at 24 months or a continuous course with relapses and remission (Ravelli et al., 2010).
Attempts to predict JDM outcomes at diagnosis with modeling have been proposed, but so far well-validated methods are lacking (Reed et al., 2019). The presence or absence of myositis-associated or -specific antibodies, muscle biopsy pathology, and organ involvement may portend a more favorable or a more severe disease course, although additional research needs to be done to validate their use (Deakin et al., 2016; Mamyrova et al., 2018; Pachman and Khojah, 2018). Without such tools, it is difficult to predict outcomes.
Treatments
There are a variety of pharmacologic treatments available for children which mirror the treatments available for adult forms of the disease. The goals for treatment of JDM are to quell the inflammation, limit irreparable damage due to the disease, and, hopefully, induce a long-term remission allowing for the potential withdrawal of medications at some point. The treatment is determined by the severity of the disease and the organ-specific involvement. Because of the heterogeneity of the disease, treatment is tailored to each specific patient, and practice variation exists on indications for each specific agent; it is impossible to describe a single approach to management.
The standard treatment usually involves corticosteroids for some period of time. Methotrexate is the most commonly used immunosuppressive medication in JDM (Hasija et al., 2011; Huber, 2018; Stringer et al., 2010). While hydroxychloroquine is often used as an adjunct to help skin disease and possibly decrease the total amount of steroids required, it is sometimes used as the sole treatment in mild cases of skin-predominant disease (Kim et al., 2017). However, it has been shown that the majority of patients with clinically amyopathic disease will not go into disease remission with antimalarial treatment alone (Pinard et al., 2019).
IVIG is the most commonly used medication in JDM after GCs, methotrexate, and hydroxychloroquine. There are differing opinions concerning the indications for IVIG, with some clinicians reserving it for patients who have more than mild myositis, others using it for recalcitrant skin disease, and others for steroid resistant disease (Lam et al., 2011). IVIG efficacy in JDM is well established (Al-Mayouf et al., 2000). Beyond these medications, a variety of immune modulation is used, including mycophenolate, azathioprine, abatacept, cyclosporine, tacrolimus, anti-B cell therapy (most commonly rituximab), anti-TNF therapy, cyclophosphamide, and JAK
inhibitors (Enders et al., 2017; Huber et al., 2012; Le Voyer et al., 2021; Stringer et al., 2010) for recalcitrant disease. The duration of therapy varies, but in a typical patient GCs may be tapered over 6 to 12 months (Huber et al., 2012) with steroid-sparing agents continued for several years. A transition from GCs to a steroid-sparing medication is typically attempted as soon as possible. Children who are not able to transition off GCs tend to have a higher risk of treatment-related complications (Martin et al., 2012).
IVIG is often used for a much shorter period of time than other treatments. The treatment duration is typically several months, until clinical improvement occurs. It is then discontinued, with the other immune-modulating agents continued for several years. In children with a chronic course, the treatment may be ongoing. Finally, it is sometimes necessary to escalate therapy specifically to address skin calcinosis, which is more likely to occur in DM than in PM, and more likely in JDM than in adult DM. In addition to the above therapies, sodium thiosulfate and bisphosphonates are also sometimes used to treat calcinosis.
Likelihood of Improvement Given Treatment
JDM historically carried a 30 percent mortality rate, which has fallen below 5 percent with modern therapy (Ravelli et al., 2010). Approximately 25 percent of patients will achieve disease inactivity and cessation of medication within 2 years of diagnosis. Another 25 percent will experience alternating periods of disease activity and remission. The remaining 50 percent of patients will have a more chronic disease course which requires ongoing pharmacologic intervention to control their symptoms (Huber, 2018). Despite the low mortality rates and the ability to achieve disease inactivity in children, morbidity can still be significant. Patients may be left with permanent reduction in muscle mass and endurance as well as skin damage (calcinosis or lipodystrophy) that will continue to affect their quality of life (Ravelli et al., 2010).
Secondary Impairments from Treatment
One unique pediatric consideration is the impact of chronic GC exposure on growth. This can lead to short stature and pubertal delay, which can in turn have significant psychosocial impact. Treatment may require a child to miss school on a regular basis for inpatient hospitalization or infusion center appointments. This, along with increased vigilance about infection leading to avoidance of social functions and sometimes school attendance, may affect the child’s social development, which is an important part of child and adolescent growth.
SUMMARY
IIMs, often collectively known as “myositis,” are rare diseases characterized by inflammation and weakness of the skeletal muscles, and they can also involve internal organs such as lungs, heart, and esophagus. In clinical practice, the most common myopathies are PM and DM. DM primarily affects the skin, muscles, joints, and lungs, whereas in PM, muscle weakness is a predominant feature, usually with no skin involvement. DM is also strongly associated with cancers. Juvenile myositis shares many clinical features with the adult forms of the disease, with some important differences. Notably, in JDM (the most common form of juvenile myositis), compared with adults DM, ILD and cancer are more rare, while GI, endocrine, and vascular effects are more common. Functional disability is common in individuals with myositis, particularly impaired muscle function, predominantly in the proximal and axial muscle groups, that results in functional limitations related to muscle weakness and decreased endurance with more fatigability. It is important to note that myositis can occur as a feature of a larger systemic autoimmune disease, such as systemic lupus erythematosus, whereas myositis is just one of many disease manifestations.
A professionally accepted diagnosis of myositis is based on a combination of a patient’s history, clinical examination, and laboratory tests such as creatine kinase and muscle biopsy. Additional testing may include myopathic features on EMG and the detection of MSAs. The diagnostic workup is similar in adults and children, but children frequently do not undergo a muscle biopsy when the characteristic rashes of JDM are present. In recent years, MSAs are increasingly used as additional evidence to make a diagnosis, select treatments, and predict disease course, for example by providing information about disease severity, the risk of systemic effects, and cancer. More research and standardization still needs to be done in this field.
The usual course of myositis is progressive, but it can be relapsing and remitting with periods of worsening and quiescence. The disease course can be categorized as monophasic, meaning a disease in which the patient can eventually be tapered off medication; chronic, meaning the patient continues to require medication; or polyphasic, meaning the patient experiences relapse and remission. The disease course and outcomes vary, and prognosis depends on systemic effects and multimorbidity. In cases where muscle inflammation leads to irreversible muscle atrophy and fatty replacement, permanent muscle weakness in those targeted muscle groups will persist because of permanent damage. ILD is a well-recognized complication of DM and PM and worsens the prognosis of the disease. Cardiac involvement is also associated with poor prognosis. Other potential longstanding complications
include joint deformities and contractures, which may occur more often in children. Even when the disease is controlled, many patients are left with damage such as calcinosis, muscle atrophy, and fatty replacement leading to permanent weakness and decreased endurance.
Corticosteroids with adjunctive steroid-sparing immunosuppressive therapies are recommended to treat disease activity, prevent mortality, and reduce long-term disability. While they are effective, corticosteroids lead to numerous complications, including hypertension, cataracts, osteoporosis, weight gain, and psychosis. In JDM, chronic GC exposure can have an impact on a child’s growth, leading to short stature and pubertal delay as well as to significant psychosocial impact. Combinations of second-line therapies or newer third-line therapies are used in severe, refractory, or corticosteroid-dependent diseases. The treatments should be chosen according to the patient’s clinical features and systemic involvement. Future trials may consider subgrouping patients into clinical and serological subtypes to help identify biomarkers for response to specific immunosuppressive and biological agents.
Prompt diagnosis and treatment generally leads to better outcomes, decreased permanent damage, and decreased long-term disability. In adults, GC treatment is able to reduce muscular inflammation, and more than 60 percent of the patients show improvement of muscle symptoms. In children, approximately 25 percent will achieve disease inactivity and cessation of medication within 24 months of diagnosis, another 25 percent will experience alternating periods of disease activity and remission, and the remaining 50 percent of patients will have a more chronic disease course requiring ongoing pharmacologic intervention to control the symptoms. Despite the ability to achieve disease inactivity, morbidity can be significant, and permanent reduction in muscle mass and skin damage can continue to affect quality of life.
REFERENCES
ACR (American College of Rheumatology). 2021. ACR image library. https://www.rheumatology.org/Learning-Center/Rheumatology-Image-Library (accessed November 15, 2021).
Aggarwal, R., L. G. Rider, N. Ruperto, N. Bayat, B. Erman, B. M. Feldman, C. V. Oddis, A. A. Amato, H. Chinoy, R. G. Cooper, M. Dastmalchi, D. Fiorentino, D. Isenberg, J. D. Katz, A. Mammen, M. de Visser, S. R. Ytterberg, I. E. Lundberg, L. Chung, K. Danko, I. Garcia-De la Torre, Y. W. Song, L. Villa, M. Rinaldi, H. Rockette, P. A. Lachenbruch, F. W. Miller, J. Vencovsky, the International Myositis Assessment and Clinical Studies Group, and the Paediatric Rheumatology International Trials Organization. 2017. 2016 American College of Rheumatology/European League Against Rheumatism criteria for minimal, moderate, and major clinical response in adult dermatomyositis and polymyositis: An International Myositis Assessment and Clinical Studies Group/Paediatric Rheumatology International Trials Organisation collaborative initiative. Annals of the Rheumatic Diseases 76(5):792–801.
Aggarwal, R., G. Marder, D. C. Koontz, P. Nandkumar, Z. Qi, and C. V. Oddis. 2018. Efficacy and safety of adrenocorticotropic hormone gel in refractory dermatomyositis and polymyositis. Annals of the Rheumatic Diseases 77(5):720–727.
Aggarwal, R., C. Charles-Schoeman, J. Schessl, M. M. Dimachkie, I. Beckmann, and T. Levine. 2021. Prospective, double-blind, randomized, placebo-controlled phase III study evaluating efficacy and safety of Octagam 10% in patients with dermatomyositis (“Proderm study”). Medicine 100(1):e23677.
Al-Mayouf, S. M., R. M. Laxer, R. Schneider, E. D. Silverman, and B. M. Feldman. 2000. Intravenous immunoglobulin therapy for juvenile dermatomyositis: Efficacy and safety. Journal of Rheumatology 27(10):2498–2503.
Albayda, J., L. R. Hayes, and L. Christopher-Stine. 2021. Dancing muscles: The value of real-time ultrasound evaluation of muscle in myositis and mimics. Rheumatology (Oxford, England) 60(8):e275–e276.
Alexanderson, H., M. Del Grande, C. O. Bingham III, A. M. Orbai, C. Sarver, K. Clegg-Smith, I. E. Lundberg, Y. W. Song, and L. Christopher-Stine. 2014. Patient-reported outcomes and adult patients’ disease experience in the idiopathic inflammatory myopathies. Report from the OMERACT 11 myositis special interest group. Journal of Rheumatology 41(3):581–592.
Amato, A. A., and R. J. Barohn. 2009. Inclusion body myositis: Old and new concepts. Journal of Neurology, Neurosurgery and Psychiatry 80(11):1186–1193.
Amici, D. R., I. Pinal-Fernandez, R. Pagkatipunan, A. Mears, R. de Lorenzo, E. Tiniakou, J. Albayda, J. J. Paik, T. E. Lloyd, L. Christopher-Stine, A. L. Mammen, and T. Chung. 2019. Muscle endurance deficits in myositis patients despite normal manual muscle testing scores. Muscle and Nerve 59(1):70–75.
Aussy, A., O. Boyer, and N. Cordel. 2017. Dermatomyositis and immune-mediated necrotizing myopathies: A window on autoimmunity and cancer. Frontiers in Immunology 8:992.
Barba, T., R. Fort, V. Cottin, S. Provencher, I. Durieu, S. Jardel, A. Hot, Q. Reynaud, and J. C. Lega. 2019. Treatment of idiopathic inflammatory myositis associated interstitial lung disease: A systematic review and meta-analysis. Autoimmunity Reviews 18(2):113–122.
Barsotti, S., and I. E. Lundberg. 2018. Myositis an evolving spectrum of disease. Immunological Medicine 41(2):46–54.
Bohan, A., and J. B. Peter. 1975. Polymyositis and dermatomyositis. The New England Journal of Medicine 292(7):344–347.
Bunch, T. W. 1981. Prednisone and azathioprine for polymyositis. Long-term followup. Arthritis & Rheumatology 24(1):45–48.
Bunch, T. W., J. W. Worthington, J. J. Combs, D. M. Ilstrup, and A. G. Engel. 1980. Azathioprine with prednisone for polymyositis. A controlled, clinical trial. Annals of Internal Medicine 92(3):365–369.
Cassius, C., H. Le Buanec, J. D. Bouaziz, and R. Amode. 2019. Biomarkers in adult dermatomyositis: Tools to help the diagnosis and predict the clinical outcome. Journal of Immunology Research 2019:9141420.
Dalakas, M. C. 2020. Inflammatory myopathies: Update on diagnosis, pathogenesis and therapies, and COVID-19-related implications. Acta Myologica 39(4):289–301.
Dalakas, M. C., I. Illa, J. M. Dambrosia, S. A. Soueidan, D. P. Stein, C. Otero, S. T. Dinsmore, and S. McCrosky. 1993. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. New England Journal of Medicine 329(27):1993–2000.
Deakin, C. T., S. A. Yasin, S. Simou, K. A. Arnold, S. L. Tansley, Z. E. Betteridge, N. J. McHugh, H. Varsani, J. L. Holton, T. S. Jacques, C. A. Pilkington, K. Nistala, L. R. Wedderburn, K. Armon, J. Ellis-Gage, H. Roper, V. Briggs, J. Watts, L. McCann, I. Roberts, E. Baildam, L. Hanna, O. Lloyd, S. Wadeson, P. Riley, A. McGovern, C. Ryder, J. Scott, B. Thomas, T. Southwood, E. Al-Abadi, S. Wyatt, G. Jackson, T. Amin, M. Wood, V. VanRooyen, D. Burton, J. Davidson, J. Gardner-Medwin, N. Martin, S. Ferguson, L. Waxman, M. Browne, M. Friswell, A. Swift, S. Jandial, V. Stevenson, D. Wade, E. Sen, E. Smith, L. Qiao, S. Watson, C. Duong, H. Venning, R. Satyapal, E. Stretton, M. Jordan, E. Mosley, A. Frost, L. Crate, K. Warrier, S. Stafford, N. Hasson, S. Maillard, E. Halkon, V. Brown, A. Juggins, S. Smith, S. Lunt, E. Enayat, L. Kassoumeri, L. Beard, Y. Glackin, B. Almeida, R. Marques, S. Dowle, C. Papadopoulou, K. Murray, J. Ioannou, L. Suffield, M. Al-Obaidi, H. Lee, S. Leach, H. Smith, A. M. McMahon, H. Chisem, R. Kingshott, N. Wilkinson, E. Inness, E. Kendall, D. Mayers, R. Etherton, K. Bailey, J. Clinch, N. Fineman, H. Pluess-Hall, L. Vallance, L. Akeroyd, A. Leahy, A. Collier, R. Cutts, H. De Graaf, B. Davidson, S. Hartfree, D. Pratt, on behalf of the UK Juvenile Dermatomyositis Research Group. 2016. Muscle biopsy findings in combination with myositis-specific autoantibodies aid prediction of outcomes in juvenile dermatomyositis. Arthritis & Rheumatology 68(11):2806–2816.
DeWane, M. E., R. Waldman, and J. Lu. 2020. Dermatomyositis: Clinical features and pathogenesis. Journal of the American Academy of Dermatology 82(2):267–281.
Enders, F. B., B. Bader-Meunier, E. Baildam, T. Constantin, P. Dolezalova, B. M. Feldman, P. Lahdenne, B. Magnusson, K. Nistala, S. Ozen, C. Pilkington, A. Ravelli, R. Russo, Y. Uziel, M. van Brussel, J. van der Net, S. Vastert, L. R. Wedderburn, N. Wulffraat, L. J. McCann, and A. van Royen-Kerkhof. 2017. Consensus-based recommendations for the management of juvenile dermatomyositis. Annals of the Rheumatic Diseases 76(2):329–340.
Espinosa-Ortega, H. F., M. Moreno-Ramirez, and H. Alexanderson. 2017. Novel insights of disability assessment in adult myositis. Current Opinion in Rheumatology 29(6):591–597.
Feldman, B. M., L. G. Rider, A. M. Reed, and L. M. Pachman. 2008. Juvenile dermatomyositis and other idiopathic inflammatory myopathies of childhood. The Lancet 371(9631):2201–2212.
Findlay, A. R., N. A. Goyal, and T. Mozaffar. 2015. An overview of polymyositis and dermatomyositis. Muscle and Nerve 51(5):638–656.
Furst, D. E., A. A. Amato, S. R. Iorga, K. Gajria, and A. W. Fernandes. 2012. Epidemiology of adult idiopathic inflammatory myopathies in a U.S. managed care plan. Muscle and Nerve 45(5):676–683.
Gerami, P., H. W. Walling, J. Lewis, L. Doughty, and R. D. Sontheimer. 2007. A systematic review of juvenile-onset clinically amyopathic dermatomyositis. British Journal of Dermatology 157(4):637–644.
Glaubitz, S., R. Zeng, and J. Schmidt. 2020. New insights into the treatment of myositis. Therapeutic Advances in Musculoskeletal Disease 12:1759720X19886494.
Gordon, P. A., J. B. Winer, J. E. Hoogendijk, and E. H. Choy. 2012. Immunosuppressant and immunomodulatory treatment for dermatomyositis and polymyositis. Cochrane Database of Systematic Reviews (Online) 8:CD003643.
Griger, Z., M. Nagy-Vincze, and K. Danko. 2017. Pharmacological management of dermatomyositis. Expert Reviews in Clinical Pharmacology 10(10):1109–1118.
Hasija, R., A. Pistorio, A. Ravelli, E. Demirkaya, R. Khubchandani, D. Guseinova, C. Malattia, H. Canhao, L. Harel, D. Foell, C. Wouters, C. De Cunto, C. Huemer, Y. Kimura, H. Mangge, C. Minetti, E. B. Nordal, P. Philippet, R. Garozzo, A. Martini, and N. Ruperto. 2011. Therapeutic approaches in the treatment of juvenile dermatomyositis in patients with recent-onset disease and in those experiencing disease flare: An international multicenter PRINTO study. Arthritis & Rheumatology 63(10):3142–3152.
Hoeltzel, M. F., E. J. Oberle, A. B Robinson, A. Agarwal, and L.G. Rider (2014). The presentation, assessment, pathogenesis, and treatment of calcinosis in juvenile dermatomyositis. Current Rheumatology Reports 16(12):467.
Huber, A. M. 2018. Juvenile idiopathic inflammatory myopathies. Pediatric Clinics of North America 65(4):739–756.
Huber, A. M., J. E. Hicks, P. A. Lachenbruch, M. D. Perez, L. S. Zemel, R. M. Rennebohm, C. A. Wallace, C. B. Lindsley, M. H. Passo, S. H. Ballinger, S. L. Bowyer, A. M. Reed, P. H. White, I. M. Katona, F. W. Miller, L. G. Rider, and B. M. Feldman. 2001. Validation of the Childhood Health Assessment Questionnaire in the juvenile idiopathic myopathies. Journal of Rheumatology 28(5):1106–1111.
Huber, A. K., F. D. Finkelman, C. W. Li, E. Concepcion, E. Smith, E. Jacobson, R. Latif, M. Keddache, W. Zhang, and Y. Tomer. 2012. Genetically driven target tissue over-expression of cd40: A novel mechanism in autoimmune disease. Journal of Immunology 189(6):3043–3053.
Kim, S., P. Kahn, A. B. Robinson, B. Lang, A. Shulman, E. J. Oberle, K. Schikler, M. L. Curran, L. Barillas-Arias, C. H. Spencer, L. G. Rider, and A. M. Huber. 2017. Childhood Arthritis and Rheumatology Research Alliance consensus clinical treatment plans for juvenile dermatomyositis with skin predominant disease. Pediatric Rheumatology 15(1):1.
Lam, C. G., C. Manlhiot, E. M. Pullenayegum, and B. M. Feldman. 2011. Efficacy of intravenous IG therapy in juvenile dermatomyositis. Annals of the Rheumatic Diseases 70(12):2089–2094.
Le Voyer, T., C. Gitiaux, F. J. Authier, C. Bodemer, I. Melki, P. Quartier, F. Aeschlimann, A. Isapof, J. P. Herbeuval, V. Bondet, J. L. Charuel, M. L. Frémond, D. Duffy, M. P. Rodero, and B. Bader-Meunier. 2021. JAK inhibitors are effective in a subset of patients with juvenile dermatomyositis: A monocentric retrospective study. Rheumatology (United Kingdom) 60(12):5801–5808.
Liao, A. P., M. Salajegheh, R. Nazareno, J. C. Kagan, R. G. Jubin, and S. A. Greenberg. 2011. Interferon β is associated with type 1 interferon-inducible gene expression in dermatomyositis. Annals of the Rheumatic Diseases 70(5):831–836.
Lilleker, J. B., and H. Chinoy. 2020. Can machine learning unravel the complex IIM spectrum? Nature Reviews Rheumatology 16(6):299–300.
Lundberg, I. E., A. Tjarnlund, M. Bottai, V. P. Werth, C. Pilkington, M. Visser, L. Alfredsson, A. A. Amato, R. J. Barohn, M. H. Liang, J. A. Singh, R. Aggarwal, S. Arnardottir, H. Chinoy, R. G. Cooper, K. Danko, M. M. Dimachkie, B. M. Feldman, I. G. Torre, P. Gordon, T. Hayashi, J. D. Katz, H. Kohsaka, P. A. Lachenbruch, B. A. Lang, Y. Li, C. V. Oddis, M. Olesinska, A. M. Reed, L. Rutkowska-Sak, H. Sanner, A. Selva-O’Callaghan, Y. W. Song, J. Vencovsky, S. R. Ytterberg, F. W. Miller, L. G. Rider, International Myositis Classification Criteria Project Consortium, the Euromyositis Register, and The Juvenile Dermatomyositis Cohort Biomarker Study and Repository (JDRC) (UK and Ireland). 2017. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Annals of Rheumatic Diseases 76(12):1955–1964.
Mamyrova, G., T. Kishi, I. N. Targoff, A. Ehrlich, R. V. Curiel, L. G. Rider, L. S. Abramson, B. Arabshahi, V. Cartwright, E. J. Chalom, B. A. Eberhardt, W. Hannan, G. C. Higgins, R. C. Fuhlbrigge, J. C. Jacobs, L. Jung, Y. Kimura, C. B. Lindsley, A. L. Martin, F. W. Miller, D. Milojevic, B. E. Ostrov, M. D. Perez, R. F. Rivas-Chacon, M. Rosenkranz, D. D. Sherry, J. Soep, S. Sule, and S. A. Vogelgesang. 2018. Features distinguishing clinically amyopathic juvenile dermatomyositis from juvenile dermatomyositis. Rheumatology (United Kingdom) 57(11):1956–1963.
Mamyrova, G., T. Kishi, M. Shi, I. N. Targoff, A. M. Huber, R. V. Curiel, F. W. Miller, and L. G. Rider. 2021. Anti-MDA5 autoantibodies associated with juvenile dermatomyositis constitute a distinct phenotype in North America. Rheumatology (Bulgaria) 60(4):1839–1849.
Martin, N., P. Krol, S. Smith, L. Beard, C. A. Pilkington, J. Davidson, and L. R. Wedderburn. 2012. Comparison of children with onset of juvenile dermatomyositis symptoms before or after their fifth birthday in a UK and Ireland juvenile dermatomyositis cohort study. Arthritis Care & Research 64(11):1665–1672.
Marvi, U., L. Chung, and D. F. Fiorentino. 2012. Clinical presentation and evaluation of dermatomyositis. Indian Journal of Dermatology 57(5):375–381.
McGrath, E. R., C. T. Doughty, and A. A. Amato. 2018. Autoimmune myopathies: Updates on evaluation and treatment. Neurotherapeutics 15(4):976–994.
Meyer, A., N. Meyer, M. Schaeffer, J. E. Gottenberg, B. Geny, and J. Sibilia. (2015). Incidence and prevalence of inflammatory myopathies: A systematic review. Rheumatology (Oxford, England) 54(1):50–63.
Meyer, A., C. A. Scire, R. Talarico, T. Alexander, Z. Amoura, T. Avcin, S. Barsotti, L. Beretta, J. Blagojevic, G. Burmester, I. Cavazzana, P. Cherrin, L. Damian, A. Doria, J. E. Fonseca, F. Furini, I. Galetti, F. Houssiau, T. Krieg, L. Maddalena, D. Launay, R. Campanilho-Marques, T. Martin, M. Matucci-Cerinic, P. Moinzadeh, C. Montecucco, M. F. Moraes-Fontes, L. Mouthon, R. Neri, S. Paolino, Y. Piette, S. Rednic, F. Tamirou, A. Tincani, N. Toplak, S. Bombardieri, E. Hachulla, U. Mueller-Ladner, M. Schneider, V. Smith, A. Vieira, M. Cutolo, M. Mosca, and L. Cavagna. 2019. Idiopathic inflammatory myopathies: State of the art on clinical practice guidelines [corrected]. RMD Open 4(Suppl 1):e000784.
Miller, F. W. 2021. Slicing and dicing myositis for cures and prevention. Nature Reviews Rheumatology 17(5):255–256.
Morris, P., and J. Dare. 2010. Juvenile dermatomyositis as a paraneoplastic phenomenon: An update. Journal of Pediatric Hematology/Oncology 32(3):189–191.
Muscle Study Group. 2011. A randomized, pilot trial of etanercept in dermatomyositis. Annals of Neurology 70(3):427–436.
Oddis, C. V., and R. Aggarwal. 2018. Treatment in myositis. Nature Reviews Rheumatology 14(5):279–289.
Oddis, C. V., A. M. Reed, R. Aggarwal, L. G. Rider, D. P. Ascherman, M. C. Levesque, R. J. Barohn, B. M. Feldman, M. O. Harris-Love, D. C. Koontz, N. Fertig, S. S. Kelley, S. L. Pryber, F. W. Miller, and H. E. Rockette. 2013. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: A randomized, placebo-phase trial. Arthritis & Rheumatology 65(2):314–324.
Ogawa-Momohara, M., Y. Muro, M. Kono, and M. Akiyama. 2019. Prognosis of dysphagia in dermatomyositis. Clinical and Experimental Rheumatology 37(1):165.
Oldroyd, A. G. S., A. B. Allard, J. P. Callen, H. Chinoy, L. Chung, D. Fiorentino, M. D. George, P. Gordon, K. Kolstad, D. J. B. Kurtzman, P. M. Machado, N. J. McHugh, A. Postolova, A. Selva-O’Callaghan, J. Schmidt, S. Tansley, R. A. Vleugels, V. P. Werth, and R. Aggarwal. 2021. A systematic review and meta-analysis to inform cancer screening guidelines in idiopathic inflammatory myopathies. Rheumatology (Oxford) 60(6):2615–2628.
Pachman, L. M., and A. M. Khojah. 2018. Advances in juvenile dermatomyositis: Myositis specific antibodies aid in understanding disease heterogeneity. Journal of Pediatrics 195:16–27.
Paik, J. J., L. Casciola-Rosen, J. Y. Shin, J. Albayda, E. Tiniakou, D. Leung, L. GutierrezAlamillo, J. Perin, L. Florea, C. Antonescu, S. G. Leung, G. Purwin, A. Koenig, and L. Christopher-Stine. 2021. Study of tofacitinib in refractory dermatomyositis: An open-label pilot study of ten patients. Arthritis & Rheumatology 73(5):858–865.
Pestronk, A. 2011. Acquired immune and inflammatory myopathies: Pathologic classification. Current Opinion in Rheumatology 23(6):595–604.
Pinal-Fernandez, I., D. R. Amici, C. A. Parks, A. Derfoul, M. Casal-Dominguez, K. Pak, R. Yeker, P. Plotz, J. C. Milisenda, J. M. Grau-Junyent, A. Selva-O’Callaghan, J. J. Paik, J. Albayda, A. M. Corse, T. E. Lloyd, L. Christopher-Stine, and A. L. Mammen. 2019. Myositis autoantigen expression correlates with muscle regeneration but not autoantibody specificity. Arthritis & Rheumatology 71(8):1371–1376.
Pinard, J., A. N. Femia, M. Roman, A. Alsarheed, C. Joyce, J. Lin, and R. A. Vleugels. 2019. Systemic treatment for clinically amyopathic dermatomyositis at 4 tertiary care centers. JAMA Dermatology 155(4):494–496.
Qiang, J. K., W. B. Kim, A. Baibergenova, and R. Alhusayen. 2017. Risk of malignancy in dermatomyositis and polymyositis. Journal of Cutaneous Medicine and Surgery 21(2):131–136.
Ramanan, A. V., and B. M. Feldman. 2002. Clinical outcomes in juvenile dermatomyositis. Current Opinion in Rheumatology 14(6):658–662.
Ravelli, A., L. Trail, C. Ferrari, N. Ruperto, A. Pistorio, C. Pilkington, S. Maillard, S. K. Oliveira, F. Sztajnbok, R. Cuttica, M. Beltramelli, F. Corona, M. M. Katsicas, R. Russo, V. Ferriani, R. Burgos-Vargas, S. Magni-Manzoni, E. Solis-Vallejo, M. Bandeira, F. Zulian, V. Baca, E. Cortis, F. Falcini, M. Alessio, M. G. Alpigiani, V. Gerloni, C. Saad-Magalhaes, R. Podda, C. A. Silva, L. Lepore, E. Felici, F. Rossi, E. Sala, and A. Martini. 2010. Long-term outcome and prognostic factors of juvenile dermatomyositis: A multinational, multicenter study of 490 patients. Arthritis Care & Research 62(1):63–72.
Redondo-Benito, A., A. Curran, A. Villar-Gomez, E. Trallero-Araguas, A. Fernández-Codina, I. Pinal-Fernandez, J. Á. Rodrigo-Pendás, and A. Selva-O’Callaghan. 2018. Opportunistic infections in patients with idiopathic inflammatory myopathies. International Journal of Rheumatic Diseases 21(2):487–496.
Reed, A. M., C. S. Crowson, and J. A. Dvergsten. 2019. A path to prediction of outcomes in juvenile idiopathic inflammatory myopathy. Frontiers in Immunology 10:638.
Rider, L. G., and K. Nistala. 2016. The juvenile idiopathic inflammatory myopathies: Pathogenesis, clinical and autoantibody phenotypes, and outcomes. Journal of Internal Medicine 280(1):24–38.
Rider, L. G., V. P. Werth, A. M. Huber, H. Alexanderson, A. P. Rao, N. Ruperto, L. Herbelin, R. Barohn, D. Isenberg, and F. W. Miller. 2011. Measures of adult and juvenile dermatomyositis, polymyositis, and inclusion body myositis. Arthritis Care & Research 63(Suppl 11):S118–S157.
Rider, L. G., R. Aggarwal, P. M. MacHado, J. Y. Hogrel, A. M. Reed, L. Christopher-Stine, and N. Ruperto. 2018. Update on outcome assessment in myositis. Nature Reviews Rheumatology 14(5):303–318.
Robinson, A. B., M. F. Hoeltzel, D. M. Wahezi, M. L. Becker, E. A. Kessler, H. Schmeling, R. Carrasco, A. M. Huber, B. M. Feldman, and A. M. Reed. 2014. Clinical characteristics of children with juvenile dermatomyositis: The Childhood Arthritis and Rheumatology Research Alliance Registry. Arthritis Care & Research 66(3):404–410.
Senécal, J. L., J. P. Raynauld, and Y. Troyanov. 2017. Editorial: A new classification of adult autoimmune myositis. Arthritis & Rheumatology 69(5):878–884.
Sizemore, T. C. 2015. In the idiopathic inflammatory myopathies, how significant is creatine kinase levels in diagnosis and progress? A case study and review of the literature. Journal of Rheumatic Diseases and Treatment 1(4).
Stringer, E., J. Bohnsack, S. L. Bowyer, T. A. Griffin, A. M. Huber, B. Lang, C. B. Lindsley, S. Ota, C. Pilkington, A. M. Reed, R. Scuccimarri, and B. M. Feldman. 2010. Treatment approaches to juvenile dermatomyositis (JDM) across North America: The Childhood Arthritis and Rheumatology Research Alliance (CARRA) JDM Treatment Survey. Journal of Rheumatology 37(9):1953–1961.
Sun, J., Y. Zheng, M. A. A. Mamun, X. Li, X. Chen, and Y. Gao. 2020. Research progress of PD-1/PD-L1 immunotherapy in gastrointestinal tumors. Biomedicine and Pharmacotherapy 129:110504.
Tjärnlund, A., Q. Tang, C. Wick, M. Dastmalchi, H. Mann, J. T. Studýnková, R. Chura, N. J. Gullick, R. Salerno, J. Rönnelid, H. Alexanderson, E. Lindroos, R. Aggarwal, P. Gordon, J. Vencovsky, and I. E. Lundberg. 2018. Abatacept in the treatment of adult dermatomyositis and polymyositis: A randomised, phase IIb treatment delayed-start trial. Annals of the Rheumatic Diseases 77(1):55–62.
Tran, T. N., A. Steffey, and H. Caspard. 2013. FRI0447 epidemiology of idiopathic inflammatory myopathies in England – a database analysis. Annals of the Rheumatic Diseases 71(Suppl 3):465–466.
Waldman, R., M. E. DeWane, and J. Lu. 2020. Dermatomyositis: Diagnosis and treatment. Journal of the American Academy of Dermatology 82(2):283–296.
Yang, Y. Y., X. X. Zuo, H. L. Zhu, and S. J. Liu. 2019. Advances in epigenetic markers of dermatomyositis/polymyositis. Beijing da xue xue bao. Yi xue ban (Journal of Peking University. Health Sciences) 51(2):374–377.






























