

The Food and Drug Administration’s Accelerated Approval Process for New Pharmaceuticals
Proceedings of a Workshop—in Brief
On January 30–31, 2023, an ad hoc committee under the auspices of the National Academies of Sciences, Engineering, and Medicine’s Committee on Science, Technology, and Law (CSTL) hosted a virtual workshop, The Food and Drug Administration’s Accelerated Approval Process for New Pharmaceuticals. The workshop examined the U.S. Food and Drug Administration’s (FDA) accelerated approval program, one of several pathways designed to facilitate and expedite the development and review of new pharmaceuticals.1 Accelerated approval provides a process for hastening access to innovative interventions for patients with serious or life-threatening diseases for which no other options are available. There have been numerous beneficial uses of accelerated approval, such as for antiretroviral drugs to treat HIV/AIDS and imatinib to treat chronic myelogenous leukemia. However, accelerated approval has been, at times, controversial due to uncertainties related to whether pharmaceuticals approved under the accelerated approval pathway, which relies on surrogate endpoints rather than a direct measure of clinical benefit, have met the same standards for safety and efficacy as drugs approved under FDA’s traditional approval pathway and whether the pathway has been used for products for which it was not intended.
CSTL Co-chair Harold Varmus (Weill Cornell Medicine) opened the workshop by noting the urgency of addressing difficult questions like accelerated approval. He welcomed participants’ input on how to balance the “needs of patients, desires of industry, and the regulatory responsibilities of FDA in getting the products of an arduous process of drug development and approval into the hands of patients who need those remedies.”
Planning committee co-chair Barbara E. Bierer (Harvard Medical School and Brigham and Women’s Hospital) reiterated that FDA’s accelerated approval process was designed to allow earlier approval of drugs that treat serious illnesses and fill an unmet medical need based on a surrogate endpoint (e.g., a biomarker).2 The accelerated approval process can considerably shorten the time to FDA approval, but drugs must still be validated through confirmatory trials. If the trials do not provide
__________________
1 For the committee’s charge, workshop agenda and video, and committee and speaker biographies, visit https://www.nationalacademies.org/event/01-30-2023/the-food-and-drug-administrations-accelerated-approval-process-for-new-pharmaceuticals-a-workshop.
2 FDA defines biomarkers as “characteristics that are objectively measured as indicators of health, disease, or a response to an exposure or intervention, including therapeutic interventions.” See https://www.fda.gov/science-research/focus-areas-regulatory-science-report/focus-area-biomarkers#:~:text=Biomarkers%20are%20characteristics%20that%20are,or%20intervention%2C%20including%20therapeutic%20interventions.
![]()
confirmation of benefit, FDA has procedures to remove the drug from the market.
Planning committee co-chair William B. Schultz (Zuckerman Spaeder LLC) noted the tension between making potentially life-saving therapies more quickly available to patients and taking time to understand if the drug works as intended based on evidence that the surrogate endpoint is “reasonably likely” to predict a clinical benefit. Some maintain that certain patients “do not have time to wait” for full FDA approval and are willing to take the risk that a drug might not work. Others argue that if drugs are available too early, patients will not participate in clinical trials and it will never be known whether the drugs really work, which disadvantages patients in the future: “This is one of the most difficult issues that any regulatory agency faces, and finding the right balance has always been a challenge for policymakers and [patient] advocates.”
OVERVIEW OF THE ACCELERATED APPROVAL PROGRAM
Session moderator and planning committee member William Hubbard (independent consultant and former FDA Associate Commissioner) provided a history of the accelerated approval program. He noted that the federal government had no authority over drug manufacturing until 1906, and initial authority was weak. In 1938, Congress enacted the Food, Drug, and Cosmetic Act, which required manufacturers to show that a drug was safe before it could be marketed. In 1962, the Act was amended to add an effectiveness standard as a requirement for FDA approval. FDA was criticized, however, for the length of time it took to approve new drugs, especially compared to other countries. In the 1970s, the National Cancer Institute, Health Care Financing Administration (HCFA)—now the Centers for Medicare and Medicaid Services (CMS)—and FDA collaborated to make some pioneering oncology drugs, known as Group C drugs, more widely available.3 In 1987, given the urgency of the HIV/AIDS epidemic and lack of available treatments, FDA approved AZT based on a surrogate endpoint.4 In 1992, based on the HIV/AIDS experience, FDA issued a final rule that set forth an accelerated approval pathway for pharmaceuticals intended to treat serious and life-threatening conditions.5 Accelerated approval was codified into statute in 2012. FDA has granted accelerated approval for about 300 drugs between 1992 and 2021.6 Hubbard called the program an overall success but noted concerns about delays in completing the confirmatory trials required for drugs approved under the program and slow withdrawals of drugs that fail to demonstrate clinical benefit (which critics say has weakened FDA’s efficacy standard).7 Most recently, he said, FDA’s controversial approval of aducanumab for treating Alzheimer’s Disease and a subsequent refusal by CMS to reimburse the full cost of the drug cast a spotlight on the program.8
The workshop’s first speaker, Jacqueline Corrigan-Curay (U.S. Food and Drug Administration) said that FDA’s statutory authority to approve a drug for sale requires substantial evidence of effectiveness and demonstration that benefits outweigh risks for the intended use.9 Accelerated approval streamlines the drug approval process by approving pharmaceuticals on the basis of their effect on surrogate endpoints that are reasonably likely to predict clinical benefit “or on a clinical endpoint” (an outcome that represents direct clinical benefit, such as survival, decreased pain, or the
__________________
3 Federal health officials recognized that cancer drugs were being developed that had not undergone the full clinical testing required for FDA approval, but that showed evidence of relative and reproducible efficacy in specific tumor types. This led FDA to develop the Group C program to distribute investigational agents to oncologists for the treatment of cancer under protocols outside the controlled clinical trial. See https://www.fda.gov/regulatory-information/search-fda-guidance-documents/treatment-use-investigational-drugs#:~:text=Group%20C%20drugs%20are%20generally,for%20specialized%20supportive%20care%20facilities.
4 CD-4 T-cell blood counts.
5 See 21 C.F.R. §§ 314.500 et seq. & 601.40 et seq. (Dec. 11, 1992).
6 See, e.g., G. Beakes-Read et al. 2022. “Analysis of FDA’s Accelerated Approval Program Performance, December 1992–December 2021.” Therapeutic Innovation & Regulatory Science, 56:698–703. doi: 10.1007/s43441-022-00430-z. “In 2021, 28% of approved drugs went through the Accelerated Approval pathway.” See also Salib, V. November 2, 2022. “Understanding the FDA’s Expedited Approval Pathways.” Pharma News Intelligence. https://pharmanewsintel.com/features/understanding-the-fdas-expedited-approval-pathways. From 2013 through 2022, FDA’s Center for Drug Evaluation and Research (CDER) has averaged about 43 novel drug approvals per year (see https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/new-drug-therapy-approvals-2022#:~:text=CDER’s%20Annual%20Novel%20Drug%20Approvals,novel%20drug%20approvals%20per%20year)
7 See, e.g., Beakes-Read et al. for a discussion of this issue.
8 The House Committee on Oversight and Reform and the Committee on Energy and Commerce report on this decision is available at https://democrats-energycommerce.house.gov/sites/democrats.energycommerce.house.gov/files/documents/Final%20Aduhelm%20Report_12.29.22.pdf).
9 Food and Drug Safety Innovations Act of 2012 (FDASIA) (Public Law 112-144), Section 506. Full text available at https://www.govinfo.gov/content/pkg/PLAW-112publ144/pdf/PLAW-112publ144.pdf.
![]()
absence of disease) “that can be measured earlier than irreversible morbidity or mortality.” FDA maintains a list of surrogate endpoints that it has identified as predictive of clinical benefit for both accelerated and traditional approval pathways.10
Corrigan-Curay distinguished between FDA’s traditional and accelerated approval pathways, but stressed that, for both pathways, sponsors must conduct adequate and well-controlled trials as a requirement for drug approval.11 She cautioned that, “there is a tradeoff between faster drug development to meet an unmet medical need for serious, life-threatening disease and the greater uncertainty as to whether the reasonably likely surrogate does indeed predict clinical benefit.” A primary challenge, she said, is to identify the surrogates that accurately predict clinical benefit.
The number of accelerated approvals has risen over time and oncology drugs have become the most frequently approved drugs (see Figure 1). Accelerated approvals were used for novel oncology drugs 67–83% of the time between 2016 and 2022, compared to 7–28% for novel non-oncology drugs. Most drugs submitted for accelerated approval convert to traditional approval or are withdrawn.12
Corrigan-Curay noted that changes enacted under Section 3210 of the Consolidated Appropriations Act, 2023 (P.L. 117-328),13 hereafter the 2023 Omnibus, gives FDA statutory authority to require that confirmatory trials are underway when it considers accelerated approval; provides detail regarding expedited withdrawal procedures for drugs on the market; allows FDA to specify conditions for post-approval studies; increases publicly available reporting from sponsors throughout the approval process; and establishes an Accelerated Approval Council.14 “We think accelerated approval will continue to be an important pathway,” she said.
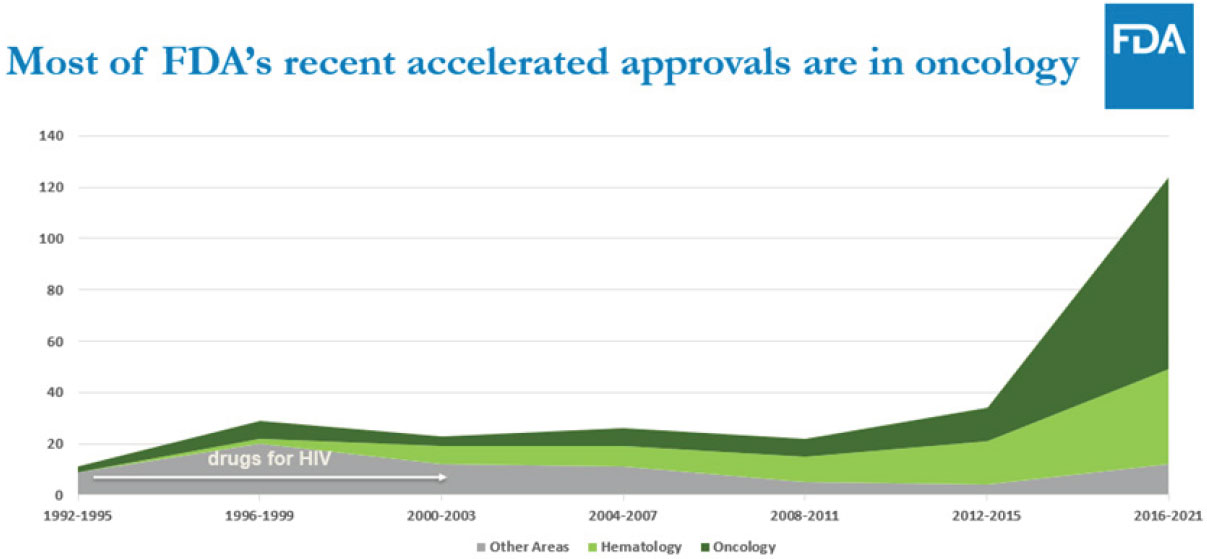
SOURCE: Jacqueline Corrigan-Curay, Workshop Presentation, January 30, 2023. Used with permission.
Former FDA Principal Deputy Commissioner Joshua Sharfstein (Johns Hopkins University) observed that stakeholder views on accelerated approval vary. For patients, he said, the question is whether the medication works, but he suggested that the accelerated approval program has resulted in mixed clinical outcomes, citing one assessment where, of the drugs examined, only one-third of oncology drugs and one-half of non-oncology drugs had high added therapeutic value.15 Critics of the program note that many patients are not likely helped by products receiving accelerated approvals, and delays in confirmatory trials affect understanding of whether many drugs work even as they remain on the market.
Sharfstein suggested that pharmaceutical companies should determine which drugs are candidates for accelerated approval at the onset of development; that FDA should clearly state which kinds of studies it will accept as confirmatory evidence, continually strive to reduce delays in the performance of confirmatory studies, strive to move quickly to withdraw products when appropriate, and increase transparency about the accelerated approval process; policymakers should establish incentives to encourage conduct of confirmatory studies; and FDA and academic experts should study the
__________________
10 See https://www.fda.gov/drugs/development-resources/table-surrogate-endpoints-were-basis-drug-approval-or-licensure.
11 FDA defines a sponsor as the company, agency, or other individual or organization that takes responsibility for and initiates a clinical investigation. Workshop speakers used terms like “industry,” “pharmaceutical companies,” and “drug developers” when referring to sponsors.
12 For a database of drugs that received accelerated approval, see https://www.fda.gov/drugs/nda-and-bla-approvals/accelerated-approval-program.
13 See https://www.congress.gov/117/bills/hr2617/BILLS-117hr2617enr.pdf for the full text of the Act.
14 Section 3210 begins on pg. 1364 of the Omnibus Act. The Council, as specified in Section 3210(e), is an intra-FDA group to “discuss issues related to accelerated approval, including any relevant cross-disciplinary approaches related to product review…”.
15 K.N. Vokinger, et al. 2022. “Therapeutic Value of Drugs Granted Accelerated Approval or Conditional Marketing Authorization in the US and Europe from 2007 to 2021.” JAMA Health Forum, 3(8): e222685. doi:10.1001/jamahealthforum.2022.2685.
![]()
accelerated approval process to better predict success for products that are considered in this pathway.
Discussion
Corrigan-Curay said FDA recognizes the difficulty in setting up randomized clinical trials (RCTs), especially for rare diseases.16 She noted that the 2023 Omnibus provides FDA the authority to push for confirmatory trials for accelerated approval, which may alter how sponsors plan when to seek accelerated approval. Hubbard and Schultz suggested that pharmaceutical companies should assume that confirmatory trials are a prerequisite for accelerated approval. Sharfstein hopes the withdrawal timeline in the 2023 Omnibus Bill gives FDA the opportunity to communicate to professional societies its clinical assessments of medications that may need withdrawal. He recommended that FDA explain the reasoning behind its decisions at all stages in the approval process. Corrigan-Curay said FDA reviews each case, including withdrawals, for lessons learned.
Peter Stein (U.S. Food and Drug Administration) acknowledged that not all drugs that receive accelerated approval will be shown to have clinical benefits, stating that, “We must be comfortable with a failure rate [for approved drugs], which is unfortunate, but the context of accelerated approval is accepting some level of uncertainty.” Corrigan-Curay added that sponsors may withdraw a drug without providing a reason, though possible reasons include drug ineffectiveness, business reasons, or the field moving on (as is common in oncology). She also noted that longer-term, neurodegenerative diseases present a challenge in designing confirmatory trials due to the lengthy period over which mental decline occurs. Bierer identified an additional challenge: FDA makes decisions at the population level and may conclude a drug should be withdrawn, but patients make individual decisions and may feel that a drug benefits them. Corrigan-Curay said when a drug is withdrawn patients have options in some situations under FDA’s expanded access program.17 Corrigan-Curay underscored that the best way to identify whether a surrogate endpoint will likely predict clinical benefit is to understand the particular disease. While there is flexibility in what FDA requires of sponsors for confirmatory trials, especially for life-threatening, rare diseases, evidence of a reasonably likely benefit is necessary. She said FDA is preparing guidance on the changes to the accelerated approval program required by the 2023 Omnibus.
SURROGATE MARKERS AND ENDPOINTS
Session moderator and planning committee Member Frank Rockhold (Duke University) introduced the session by sharing FDA’s definitions of “reasonably likely” and “validated” surrogate endpoints. Reasonably likely surrogate endpoints are “supported by strong mechanistic and/or epidemiologic rationale, but the amount of clinical data is not sufficient to show they are a validated surrogate endpoint.” Validated surrogate endpoints “are supported by a clear mechanistic rationale and clinical data providing strong evidence that an effect on the surrogate endpoint predicts a specific clinical benefit.”18
David DeMets (University of Wisconsin, Madison) emphasized that relying on surrogates may miss all effects of an intervention on clinical outcome and that correlation is not causation.19 He enumerated some “surrogate failures” where intermediate markers did not result in hoped-for clinical outcomes20 and described cases where intermediate markers indicated potentially negative outcomes but where full clinical trials demonstrated positive outcomes. While intermediate markers are important in the early phases of drug research, he said, resulting Phase III trials can yield mixed results, and “surrogates do not reliably predict clinical outcome.”21 DeMets said that clinical trials with
__________________
16 Randomized clinical trials are studies in which people are divided by chance into separate groups to compare different treatments or other interventions.
17 Expanded access, also called “compassionate use,” provides access to investigational medical products under certain conditions (see https://www.fda.gov/news-events/public-health-focus/expanded-access).
18 See https://www.fda.gov/drugs/development-resources/surrogate-endpont-resources-drug-and-biologic-development.
19 DeMets noted he drew on an earlier paper for his presentation: T.R. Fleming and D.L. DeMets. 1996. “Surrogate Endpoints in Clinical Trials: Are We Being Misled?” Annals of Internal Medicine. doi: 10.7326/0003-4819-125-7-199610010-00011.
20 Such as the Cardiac Arrhythmia Suppression Trial, where drugs suppressed arrhythmias but increased deaths. See C. Pratt and L. Moye. 1995. “The Cardiac Arrhythmia Suppression Trial: Casting Suppression in A New Light.” Circulation, 91, pp. 245–247.
21 A Phase III trial is one in which “the experimental study drug or treatment is given to large groups of people. Researchers confirm its effectiveness, monitor side effects, compare it to commonly used treatments, and collect information that will allow the experimental drug or treatment to be used safely.” See https://
![]()
clinical (or validated) endpoints should be the favored mechanism for demonstrating efficacy: “There are situations where intermediate markers are necessary, but we must be careful not to forget what we do and do not know and be willing to take that risk.”
Peter Stein emphasized that, when surrogates are factors in traditional drug approvals, the endpoint is expected to predict clinical benefit, which he defined as “an improvement in how the patient feels, functions, or survives.” While substantial evidence is required as part of the accelerated approval process, the tradeoff for earlier access is greater uncertainty, including whether an endpoint actually predicts clinical benefit. He noted that confirmatory trials associated with accelerated approval applications may have, due to ethical and feasibility considerations, a shorter timeframe or include fewer participants than trials for traditional approvals.22 Stein said FDA considers data related to each disease in its assessments and, echoing DeMets, said correlation is insufficient evidence for selecting a surrogate.
Scott Emerson (University of Washington) focused on the interaction between surrogate endpoints and disease. As an example, he highlighted a 2021 meta-analysis of studies on cognitive decline versus amyloid reduction.23 The authors identified thirty-four clinical trials to review but only had access to fourteen, which Emerson commented was problematic and which he said the authors acknowledged could bias their analysis. Using a set of measures for the drugs analyzed, clinical trials of the drugs did not show marked improvements in cognitive decline. Emerson shared concerns about a review conducted by FDA on the association of amyloid plaque reduction and clinical outcomes that found a reasonably likely benefit from use of aducanumab.24 He said that the best approach to reviewing evidence of effectiveness is to conduct a meta-analysis across relevant classes of drugs as a group and to look at the magnitude of effects, not just correlation, to determine if the drugs are reasonably likely to lead to a clinically important difference. Emerson warned that there are many ways to “go astray,” including in cases where there is ambiguity on the measure of clinical endpoints and surrogates, potential bias in data, accounting for random effects, and linearity of associations25 that might influence interpretation of results.
Steve Pearson (Institute for Clinical and Economic Review [ICER]) spoke about the use of surrogate endpoints in health technology assessments (HTAs). Used in many countries outside the United States, HTA bodies are governmental agencies that receive information from a regulator to help governments and other users understand the comparative clinical and cost effectiveness of a therapy. ICER, though not a government agency, conducts analyses similar to an HTA. He said that drugs approved by an accelerated approval pathway often have high prices and limited competition. He said some observers have suggested that, in recent years, FDA has allowed drug developers more latitude with regard to demonstrating benefit if the drug meets an unmet need and has “limited or no safety signals.” Pearson urged FDA to produce and apply more transparent, consistent criteria related to causality, biological plausibility, specificity, proportionality, and universality,26 stating that FDA has inconsistently applied these criteria in approval decisions.27 He concluded that, “FDA has no framework for a consistent approach
__________________
22 The Federal Food, Drug and Cosmetic Act states: “the evidence to support that an endpoint is reasonably likely to predict clinical benefit…may include epidemiological, pathophysiological, therapeutic, pharmacologic, or other evidence using biomarkers, for example, or other scientific methods or tools.” See Section 356 (c), at https://www.govinfo.gov/content/pkg/USCODE-2019-title21/html/USCODE-2019-title21-chap9-subchapV-partA-sec356.htm.
23 S. Ackley et al. 2021. “Effects of Reductions in Amyloid Levels on Cognitive Change in Randomized Trials; Instrumental Variable Meta-Analysis.” BMJ, 372(156); updated BMJ, 378(o2084). https://www.bmj.com/content/372/bmj.n156. Amyloids are naturally occurring proteins, but one form, called beta-amyloid 42, has been found to occur at abnormal levels in the brains of patient with Alzheimer’s disease (See https://www.nia.nih.gov/health/what-happens-brain-alzheimers-disease).
24 FDA Center for Drug Evaluation and Research, Office of Clinical Pharmacology Review, Association of Amyloid Plaque Reduction and Clinical Outcomes, available at https://www.govinfo.gov/content/pkg/USCODE-2019-title21/html/USCODE-2019-title21-chap9-subchapVpartA-sec356.htm. Emerson commented that when he reviewed the data, he saw little difference between most of the drugs studied and a placebo.
25 The ability (within a given range) to obtain test results which are directly proportional to the concentration (amount) of an analyte (see https://www.fda.gov/media/152208/download).
26 Pearson noted these five criteria were elaborated upon in a presentation by L.M. McShane titled Concepts and Case Study Template for Surrogate Endpoints Workshop (see https://fnih.org/sites/default/files/final/pdf/6-McShane-Case%20Study%20Overview.pdf. They are: Causality; Plausibility; Specificity; Proportionality; and Universality (see slide 12).
27 Two examples Emerson cited were eteplirsen (to treat Duchenne muscular dystrophy) and pembrolizum (to treat advanced cervical cancer).
![]()
to determining thresholds for meaningful change in surrogate endpoints or for explaining its final decisions” but suggested that increased transparency would help FDA with internal calibration and public accountability, provide support for controversial decisions, and create incentives for sponsors to do rigorous science on surrogates prior to FDA submission.28
Discussion
Rockhold asked about the expectations for evidence linking reasonably likely markers and clinical outcomes. Stein said FDA looks at epidemiological, biological, and natural history information, but sometimes this does not predict clinical benefits, and crossover studies complicate the picture.29 Emerson said that lowering thresholds due to an unmet need is insufficient; instead, there should be “an unmet need with a strong avenue of investigation.” DeMets suggested reviewing 20 years of cases that used modified intermediate markers, including cases beyond those that have received accelerated approval. Emerson suggested considering confirmatory trial results and experiences in different countries to help reduce uncertainty. Stein said identifying surrogate markers for rare diseases is challenging because large epidemiolocal datasets and registries may not be available. Pearson suggested that rare diseases may have an identified pathway that makes a biomarker a reasonably likely surrogate, whereas common diseases may have many genetic pathways.30 Stein countered that a genetic defect may create an easier situation for identifying biomarkers, but also may modulate multiple pathways and affect multiple biomarkers. Pearson underscored that, “Context matters. If there is no known risk and there is average benefit, then some people are getting benefit. But if there are harms and only some patients benefit, this is a different decision.”
CONFIRMATORY TRIALS
Session moderator Bierer said that FDA requires confirmatory trials to validate correlation of a surrogate endpoint with clinical benefit. Most confirmatory trials are completed, though often with significant delays.
Joseph Ross (Yale University School of Medicine) shared research on post-marketing studies of drugs and devices. He said that such research is a challenge because FDA’s public-facing database, while updated quarterly, includes inconsistent information and FDA removes completed post-marketing studies. Ross urged the agency not to delete older studies from the database. In reviewing post-marketing requirements from 2009–2012,31 including those for accelerated approvals, Ross found that only 30% of confirmatory trials had been completed and trial results reported within five years after the approvals were granted. This increased to about two-thirds after eight years.32 “That’s a long time for residual uncertainty to persist,” he said. He suggested that “Having confirmatory studies started prior to accelerated approval is probably the most important factor in timely completion.”33
Ginny Beakes-Read (Amgen) reported that data from three 10-year periods showed, in the most recent decade, a reduction in the time to convert from accelerated to traditional approval from about 4 to 2.3 years.34 The goal of shortening the period between accelerated approval and conversion or withdrawal requires that FDA have effective management tools, she said. She urged early, iterative dialogue between industry and FDA. Beakes-Read suggested that the few formal FDA withdrawal proceedings are outliers (two of 278 approvals over 30 years), and that voluntary withdrawals and other FDA
__________________
28 See A. Katenboeck, A. Mehlman, and S.D. Pearson. 2021. “Potential Policy Reforms to Strengthen the Accelerated Review Process.” Journal of Comparative Efficiency Research, 10(16). doi: 10.2217/cer-2021-0184.
29 In a crossover study, participants may receive more than one intervention, which makes determining each intervention’s specific effect more difficult. A crossover study in the context of oncology permits a patient to receive an investigational drug after a defined period if their disease progresses, but this can complicate interpretation of whether the treatment was effective.
30 A disease pathway is the sequence of biological or biochemical events that manifest during the course of a disease. See https://ncithesaurus.nci.nih.gov/ncitbrowser/pages/search_results.jsf.
31 “Postmarketing requirements (PMRs) include studies and clinical trials that sponsors are required to conduct under one or more statutes or regulations.” See https://www.fda.gov/drugs/guidance-compliance-regulatory-information/postmarket-requirements-and-commitments.
32 J.D. Wallach et al. 2018. “Postmarket Studies Required by the US Food And Drug Administration for New Drugs and Biologics Approved Between 2009 and 2012: Cross Sectional Analysis.” BMJ, 361: k2031. doi: 10.1136/bmj.k2031. The length of time for completion of confirmatory trials is, however, dependent on numerous factors (e.g., type of study).
33 These findings are consistent with FDA’s own review, Ross noted. See M. Shahzad, et al. 2023. “Association Between Preapproval Confirmatory Trial Initiation and Conversion to Traditional Approval or Withdrawal in the FDA Accelerated Approval Pathway.” JAMA, 329(9):760-761. doi:10.1001/jama.2023.0625.
34 Beakes-Read et al.
![]()
tools can be effective. She also suggested that FDA publish timelines for confirmatory trials.
Marc Buyse (International Drug Development Institute) used the case of pathological complete response (pCR) in early operable breast cancer as “an archetypical example of an endpoint used as a surrogate without evidence of statistical validity.”35 In RCTs where patients received various treatments prior to surgery (known as neo-adjuvant therapy), their pCR (the absence of a tumor at surgery) was strongly correlated to their clinical outcomes of interest—time to disease, recurrence, and death. Yet, a meta-analysis of 12 RCTs suggests that the treatment effect on pCR is not correlated to the treatment effect on the clinical outcomes: in other words, pCR is not a statistically validated surrogate, even though it can be used for accelerated approval.36 The implication for confirmatory trials, Buyse continued, is that such trials are required to confirm long-term benefits of newly approved drugs. Expecting long-term benefits is, however, a high bar, because patients with pCR at the time of surgery may receive minimal therapy, while those without pCR may receive more aggressive therapy (thus introducing a systematic difference in treatment based on pCR).37 “We need confirmatory trials not just to confirm surrogacy, but also to show that we have not harmed patients by giving them a new drug.”
Discussion
Beakes-Read said, “If studies aren’t required, then FDA needs to publish why they’re not required.” Further, the agency should share more information on sponsors’ post-approval obligations in its public database. Ross said an external advisory group could strengthen the accelerated approval process by providing guidance on expedited withdrawals or improved study designs. Beakes-Read noted that FDA considers an entire data package: when a confirmatory trial does not indicate benefit, it could be due to drug failure, faulty study design or failed trial enrollment. Buyse said sponsors sometimes want to terminate trials, but should have a long-term obligation when harmful effects on patients manifest many years later. Ross noted that FDA approval is “a signature moment” because it allows drugs to qualify for reimbursement from CMS and private payors. Buyse said that real world evidence is rarely useful for confirming benefits because such evidence is hopelessly biased due to the small or moderate effects that are seen with most drugs.
POTENTIAL BENEFITS AND ADVERSE CONSEQUENCES
Patients, sponsors, payors, and other stakeholders have different perspectives on accelerated approvals, said session moderator and planning committee member Halima Amjad (Johns Hopkins School of Medicine), a dementia specialist. She and colleagues have explored how to weigh benefits and risks in the context of FDA’s 2021 approval of aducanumab.
John Jenkins (Greenleaf Health) said that balancing earlier access and maintenance of standards is controversial and that failures are inherent: “If FDA gets it[s approvals] right 100 percent of the time, then it is not taking the appropriate level of risk.” FDA has never defined an acceptable failure percentage, but Jenkins said that some believe 20% is an acceptable failure rate, given the option for withdrawals. While FDA has been a good steward of the accelerated approval program, he said the agency damaged its credibility with aducanumab due to a lack of transparency about its decision-making. Jenkins called for sponsors to submit clear drug development plans that include detailed information about the design, endpoints, and timing of confirmatory trials. He suggested that “the ability to conduct confirmatory trials after approval is the Achilles’ heel in the process,” and that the withdrawal process is a second “Achilles’ heel.” To maintain integrity, he said, FDA and policymakers must explore ways to expedite withdrawals while developing options for patients who say they are benefiting from the treatment.
Lee Fleisher (Centers for Medicare and Medicaid Services [CMS]) discussed how CMS makes coverage decisions,
__________________
35 Pathological complete response (pCR) is the lack of all signs of cancer in tissue samples removed during surgery or biopsy after treatment with radiation or chemotherapy.
36 P. Cortazar. 2014. “Pathological Complete Response and Long-Term Clinical Benefit in Breast Cancer: The Ctneobc Pooled Analysis.” The Lancet, 384 (9938). doi: 10.1016/S0140-6736(13)62422-8. FDA states that pCR can be used as evidence in support of accelerated approval.
37 M. Buyse et al. 2022. “Surrogacy Beyond Prognosis: The Importance of “Trial-Level” Surrogacy.” The Oncologist 27(4). doi: 10.1093/oncolo/oyac006.
![]()
as established by Congress, to determine if a drug, device, or service is “reasonable and necessary” for individuals served by Medicare. Decisions, including for accelerated approval drugs, fall under National Coverage Decisions (NCD), Local Coverage Decisions, or claim-by-claim adjudications. CMS conducts three to five NCDs annually.38 NCDs are separate from decisions regarding payment for drugs, Fleisher said, and regulations govern the timetable for making decisions and soliciting public comment. A two-part NCD was implemented for monoclonal antibodies, such as aducanumab, that target amyloids. It stated: (1) CMS will cover drugs that receive traditional approval under coverage with evidence development which could include enrollment in a registry; and (2) CMS will cover drugs that have accelerated approval under coverage with evidence development if enrolled in RCTs. Fleisher said CMS made its aducanumab decision as required by statute.
As Donna Cryer (Global Liver Institute) commented, “No one understands uncertainty better than patients.” Involvement in regulatory science, she said, has become a way for patients and patient advocates to influence decision-making. Patients “must feel equally served,” she said, underscoring the need to balance “patient-defined safety and expectations of effectiveness” with “the different pace of science and regulatory constraints.” Confirmatory trials are essential, Cryer said, as they may reveal that certain subpopulations benefit from a drug while an entire patient population may not. She called for expanding trial design by looking at new sources of data through the use of NIH registries and public-private partnerships.
Gideon Blumenthal (Merck) discussed the accelerated approval process in the context of oncology.39 With regard to surrogate endpoints, trials for oncology treatments have the advantage of the ability to measure tumors, tumor progression, and, with some limitations, correlate results with long-term endpoints such as mortality. FDA’s Office of Oncologic Diseases has improved transparency and resolved some “dangling accelerated approvals” that did not have positive confirmatory trials through its advisory committees and voluntary withdrawals, he said. And requiring that confirmatory studies are fully enrolled may work against precision medicine and populations of patients with rare diseases where RCTs may not be feasible. Industry sometimes runs large umbrella trials in which the optimal patient population is not clear until late in a drug’s development—and sponsors and FDA must adapt to this changing landscape: “Validating surrogates, response rates, and progression-free survival are good endpoints, but we have a long way to go to capture the patient’s entire journey.”
Aaron Kesselheim (Harvard University) expressed concern when accelerated approval is pursued late rather than early in drug development and about the use of surrogates that do not have a clear connection to clinical benefit. There is a need for clearer communication to patients and doctors about accelerated versus traditional approvals, he said. When confirmatory trials are not done in a timely manner, use unreasonable endpoints, or are negative, the process for withdrawing products from the market has been a problem, Kesselheim said (though he noted FDA has improved). He suggested reforms related to: (1) process, to encourage clinical endpoints for post-approval studies and a potential process to withdraw drugs with negative trials automatically; (2) information, to improve clarity and updates; and (3) payment for drugs. Kesselheim suggested that using rebates, price limitations imposed before confirmatory trials, or other measures could help indicate that accelerated approval drugs do not have the same level of certainty about their clinical effects as traditionally approved drugs.
Discussion
Kesselheim acknowledged that indication-specific pricing for both accelerated and traditional approval would add complexity but suggested that, given current drug costs, experimenting with other pricing models might be worthwhile. Blumenthal said that, as progression-free survival of cancer patients and overall response have a high, but not definitive, likelihood of showing that a drug
__________________
38 Federal Register Note, “Medicare Program; Revised Process for Making National Coverage Determinations.” See https://www.govinfo.gov/content/pkg/FR-2013-08-07/pdf/2013-19060.pdf.
39 FDA’s Project CONFIRM database provides information on oncology indications and trials (see https://www.fda.gov/about-fda/oncology-center-excellence/project-confirm).
![]()
is beneficial, it is important to improve precision around survival predictions. Jenkins said this issue highlights FDA’s challenges when looking at specific drugs and reiterated a need for greater transparency regarding FDA’s principles. Cryer urged FDA to formalize patient involvement as part of decision-making. Jenkins said FDA should consider the feasibility of a confirmatory trial as part of the approval process: options may include requiring that a trial is fully enrolled pre-approval, conducting global trials, or using real-world evidence in some situations. He noted that FDA’s guidance for drugs for nonalcoholic steatohepatitis (NASH) has two steps—the second considers clinical outcomes over years. Cryer suggested that NASH is a case where standards of care, biomarkers, and meaningful endpoints are evolving and that stopping disease progression while other treatments are developed is beneficial. She emphasized that for drugs approved for unmet needs, patients may value “something versus nothing,” adding “when there are other options [for treatment], that is a different conversation.” Discussants suggested that expanding accelerated approval to new disease areas requires careful thought and that all policies need not be rewritten based on a few outliers.
OTHER NATIONS’ APPROACHES TO ACCELERATED/CONDITIONAL DRUG APPROVAL
Planning committee member Rogério Gaspar (World Health Organization) moderated a panel that considered lessons from other nations’ approaches to accelerated/conditional drug approvals.
Yasuhiro Fujiwara (Pharmaceuticals and Medical Devices Agency [PMDA]) focused on a Japanese regulatory pathway known as conditional early approval as most comparable to the U.S. accelerated approval program. Criteria for this class of approval include disease severity and high clinical utility of the drug. Confirmatory trials are not required, but post-marketing surveillance is required to confirm safety and efficacy, as is reporting and re-examination of drugs for up to ten years. Initial approval can be revoked or modified. PDMA offers product-specific consultations with industry, academia, and start-ups and has a robust monitoring system called MID-NET. Fujiwara said Japan’s accelerated or expedited approvals are based on a healthcare framework that supports transparency, product-specific clinical practice guidelines, use in limited medical institutions, post-market safety monitoring, and post-market adverse reaction relief.
Daniel O’Connor (Medicines and Healthcare Products Regulatory Agency [MHRA]) said that, in the UK’s accelerated approval processes, the key goals are prioritizing patients and bringing medical products safely and speedily to market.40 The Early Access to Medicine Scheme (EAMS) provides two-step pre-licensing access: (1) Promising Innovative Medicine Designation; and (2) EAMS Scientific Opinion to review quality, safety, and efficacy data (so that patients with life-threatening or seriously debilitating conditions can benefit from medicines before they are formally licensed). Since 2022, a framework has been used that collects real-world evidence to maximize learning and support patient access. Other MHRA flexibilities include rolling reviews, “Day 150” (150-day timeline for high-quality marketing authorization applications [MAAs]), conditional marketing authorizations, and a program for exceptional circumstances. The Innovative Licensing and Access Pathway (ILAP), introduced in 2021, partners MHRA with three HTA bodies to align regulations and access.41 To enhance regulatory decision-making, MRHA developed a Patient Involvement Strategy to better understand patient perspectives of benefit-risk.42
Anabela Marçal (European Medicines Agency) described the European Union’s (EU) Conditional Marketing Authorization (CMA), which covers serious or life-threatening diseases, emergency situations, and orphan products.43 CMAs are renewed annually, and a CMA designation is clearly shown on product packaging. Applicants are subject to specific obligations and legal
__________________
40 For more information on MHRA, see https://www.gov.uk/government/organisations/medicines-and-healthcare-products-regulatory-agency.
41 These three bodies are the National Institute for Health and Care Excellence, Scottish Medicines Consortium, and All Wales Therapeutics and Toxicology Centre.
42 MHRA, Patient Involvement Strategy 2021-25, available at: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1022370/Patient_involvement_strategy.pdf.
43 Marçal noted that EU’s CMA is most like U.S. accelerated approval. The EU’s “accelerated assessment” has a maximum 150-day timeframe. The EMA mechanisms are specified in Article 14-a of Regulation (EC) No. 726/2004.
![]()
commitments to provide missing data. The ultimate goal is to obtain a standard authorization (which, on average, takes four years). In the few cases of noncompliance or where data do not confirm initial benefit, EMA can “vary, suspend, or revoke” authorization. EMA urges applicants to plan for and justify use of CMAs early in drug development. Marçal identified as challenges: (1) correlating surrogates and clinical endpoints; (2) identifying appropriate specific obligations; and (3) deciding if there is enough information to make clinical and other decisions.
Discussion
Marçal observed that compliance has not been a major issue in the EU, perhaps because sponsors must show progress on specific obligations at their annual renewals. Fujiwara said Japan’s system for revoking approval allows for re-examination in 4 to 10 years. O’Connor said UK MHRA has tools to ensure post-authorization data are collected but noted that regulators should understand what data may be feasibly collected and how the future landscape may change. In the UK, O’Connor said ILAP was established to ensure alignment with decisionmakers, and, in general terms, that drug prices that companies can charge reflect uncertainty in data collected through expedited pathways. Marçal explained that, while EMA authorizes products, decisions about availability and reimbursement occur at the national level. While conditional or accelerated approval is not linked to price in Japan, Fujiwara said the Ministry of Health reviews possible re-pricing of drugs every 4 years. Marçal said companies are urged to validate markers with EMA’s qualification group to facilitate assessment, but surrogate markers may not correlate with clinical endpoints, which concerns EMA. O’Connor said MHRA is considering what validated endpoints mean in different contexts. Marçal noted that slower-than-expected progress on confirmatory studies is discussed in benefit-risk reviews during annual renewals. O’Connor said MHRA recognizes the changing preferences for which clinical trials patients or investigators want to be involved in, but sometimes it must push sponsors to meet confirmatory trial commitments. Fujiwara said that politicians do not pressure PDMA, but that newspapers, advocacy groups, and patients sometimes do, and he counsels his staff to remain neutral, scientific, and transparent. Marçal called for more global collaboration to speed drug development and proposed a forum where companies could meet with regulators to understand their expectations. O’Connor highlighted MHRA’s work to define “innovative” and “unmet needs” and called for wider collaboration across the life sciences and the integration of patient voices in decision-making. Fujiwara called for greater patient and advocacy involvement, regional and international collaboration, and increased research and development by U.S. biotechnology companies in Japan.
NEW DIRECTIONS
Schultz moderated the final panel, where presenters reflected on ideas raised at the workshop and offered their own.
Pilar Ossorio (University of Wisconsin, Madision) said that FDA is in a unique position to require the release of information about medical products that society is unlikely to get otherwise. FDA should not downplay its information-enforcing function, despite the fact that accelerated approval initially requires less information. Her concern with aducanumab was that two trials led to different results, which was not taken into account by FDA. Ossorio said that patient opportunity costs if an ineffective drug remains in the market include deciding not to participate in potentially more beneficial investigational trials, financial burdens, and forgoing existing options. For society, costs include a lost opportunity to research other options. When ineffective drugs become the standard of care, she suggested, longer-term, cumulative harms are more likely: “We are trading off the interests of current [patients] against future patients.” Ossorio suggested that FDA develop guidelines to evaluate chosen surrogates and thresholds for what counts as meaningful change. FDA should publicly share reasons for departing from these guidelines, with decisions reviewed by an independent committee. FDA flexibility is needed, she said, but so are guardrails. Ossorio cautioned against claiming clinical outcomes are impossible to obtain and suggested that, in cases where companies are working on the
![]()
same endpoint, public-private partnerships to conduct independent long-term research may be useful.
Rachel Sherman (Rachel Sherman Partners LLC) said that, while seen as risky at the time, accelerated approval was a huge victory when, in the 1990s, surrogates were validated for HIV treatments. “Things got messy” when sponsors submitted applications for drugs to treat diseases that affect small groups of patients, and it was necessary to decide how to conduct confirmatory trials. For many rare diseases, confirmatory trials take years, and Sherman expressed concern about setting unrealistic deadlines for completion. While FDA appears to be on the right track, she noted an inconsistency regarding verification conditions between FDA’s existing regulations and 2019 guidance about the amount of evidence a sponsor must provide for accelerated approval.44 Sherman said that weighing risks and benefits depends on the disease, patient, and options: “When there’s nothing available, patients want access. Where there are options available, patients want data. It depends where on the spectrum of the disease and the availability of therapies the patient is.”
H.G.M. (Bert) Leufkens (Utrecht University) said medicine is a continuous process without a predictable ending. Accepting evidence too early may result in a “Type 1” error that unsafely exposes patients, but if the process moves too cautiously, a “Type 2 error” may occur, and patients cannot benefit from the therapy.45 Systems must minimize both error types. Weighing evidence means dealing with uncertainties, including the possibility that new studies may not change the picture. Requiring confirmatory trials is easy, Leufkens said, but determining whether extra study changes uncertainties is hard. In Europe, debates are underway about whether too much flexibility benefits patients and about how much data to collect. In the future, Leufkens foresees that HTAs, patients, and physicians will have a larger impact on the actual use of licensed products. To fulfill greater societal demands for transparency, he expects more pre-planning by sponsors in determining their study design endpoints and more information provided by regulators about their approval decisions. He said greater transparency requires more and earlier dialogue among regulators, sponsors, prescribers, HTAs, and other stakeholders. Regulatory systems “that are too far from the clinic and real life are not sustainable and will fail,” he concluded.
Holly Fernandez Lynch (University of Pennsylvania) suggested improving accelerated approval by utilizing payment levers, tapping untapped patient-advocate capacity, and making reforms beyond those in the 2023 Omnibus Bill (e.g., making accelerated approvals temporary by assigning a date when they will be withdrawn if they are not converted to a traditional approval). Although payment is outside of its jurisdiction, she noted that FDA has flexibility in the quantity and quality of evidence it relies on to support different types of approvals. While unvalidated or intermediate surrogate endpoints may be acceptable for accelerated approvals, she said, drug pricing should reflect this uncertainty just as uncertainty is reflected in the prices of other items in the marketplace: “Payment ought to be based on the quality of the evidence.” Adjusting prices downwards for drugs with more limited evidence should not be cast as a financial loss to companies but rather as an opportunity to get drugs on the market sooner, even if not at full price. She suggested that patient advocates push companies to complete confirmatory trials, encourage trial participation, and push back on continued use of surrogate endpoints and called for movement away from the term “accelerated approval,” which emphasizes speed rather than uncertainty and confuses clinicians and patients.
Discussion
While Ossorio thinks FDA should not be involved in pricing and that reimbursements are complex in the United States, she favors differential pricing—despite the challenges of moving to such a system. She said FDA is more qualified than payors to assess the quality of evidence, but payors have a role to play in evaluating
__________________
44 For the 2019 guidance, see https://www.fda.gov/regulatory-information/search-fda-guidance-documents/labeling-human-prescription-drug-and-biological-products-approved-under-accelerated-approval.
45 A Type 1 error is a “false positive,” i.e., the conclusion that a therapy has an effect when in fact it does not. A Type 2 error is a “false negative,” i.e., the conclusion that a therapy has no effect when in fact it does.
![]()
evidence. Leufkens noted that HTAs outside the United States are considering differential reimbursements, but practical and legal issues arise. Sherman described as “beyond dismal” the current system of providing patients with better information through labeling, informed consent, and other measures. “Either we believe that an accelerated-approval product is an approved product or [we believe that] it should be an investigational drug,” she said, and, while plans may change over time, a trial should be underway at the onset of the approval process.
CONCLUDING THOUGHTS
Members of the planning committee shared their takeaways, with Hubbard returning to “the importance of the efficacy standard that is the foundation on which patient safety, as well as drug development and industry success, are based.” He expressed concern about instances where an inappropriate use of the accelerated approval process weakens the efficacy standard and called for increased awareness about what a surrogate can support vis-à-vis predictions made about the efficacy of drugs. Gaspar noted common challenges faced by all nations, while recognizing that different approaches exist based on health care and reimbursement systems, historical background, legal frameworks, and other variables. He said countries should build on the global cooperation developed during COVID-19 and expand cooperation to include discussion of accelerated approval issues. The role of real-world evidence, better harmonization, sharing of data collection, and greater levels of patient involvement should be considered. Gaspar said that regulatory systems should not be isolated from medical settings and health systems but rather should be integrated into clinical practice. To increase reliance and cooperation across regulatory environments, he urged greater participation by FDA and other agencies in international regulatory bodies. Rockhold said that, “We don’t always have access to why failures happen. The data do not seem as transparent, but if we want to learn from the information, we need more access.” Pointing to the discussion on surrogate endpoints and markers and the fact that, while some do indicate true clinical benefits, “We cannot lose sight that they are called ‘surrogate’ for a reason.” Good diagnostics do not necessarily make good surrogate markers. Amjad agreed that accelerated approvals have largely been effective, and that failures and uncertainties are expected with such a pathway. She acknowledged needing to proceed on a case-by-case basis, but noted “the need for transparency, communication, and information throughout the process.” She noted the advantages of starting confirmatory trials pre-approval but said that innovation in trial design and data collection can provide valuable information. Amjad emphasized the importance of the patient voice when considering benefits and risks and suggested that FDA and industry learn from controversial decisions, especially for non-oncological and non-HIV drugs. Schultz identified five takeaways from the workshop: (1) better information could increase patient and physician awareness that a drug has received accelerated approval; (2) confirmatory trials are the Achilles’ heel of the accelerated approval process: after approval, patients may not remain in a trial, and FDA has to consider the risks incurred if the trial remains incomplete; (3) while there are pressures to address patient needs, FDA must conduct a risk-benefit analysis for the benefit of future patients; (4) FDA has an interest in using accelerated approval for diseases other than cancer and HIV, but there are perils, and complexities involved in withdrawing a drug; and (5) surrogates are “an area that is crying out for more standards” and there must be consistency across FDA. Bierer said that the importance of the evidentiary standard for approval must not be compromised. While sympathetic to patients with unmet medical needs in life-threatening circumstances, she said that “the worst thing [to do] is not to acknowledge what we know and don’t know,” and this requires knowledge of risks and benefits. “We want,” she said, FDA “to exercise the kind of thoughtful approach and deliberation that we have come to depend on and to only approve what they have confidence in.” Bierer said that we should be asking FDA for greater transparency regarding the data used to support agency approval decisions and suggested that industry should provide a drug development plan for accelerated approval as rigorous as plans developed for traditional approval.
![]()
DISCLAIMER This Proceedings of a Workshop—in Brief has been prepared by Paula Whitacre, Steven Kendall, Anne-Marie Mazza, and Matt Cowan as a factual summary of what occurred at the workshop. The committee’s role was limited to planning the event. The statements made are those of the individual workshop participants and do not necessarily represent the views of all participants, the project sponsors, the planning committee, the Committee on Science, Technology, and Law, or the National Academies.
REVIEWERS To ensure that it meets institutional standards for quality and objectivity, this Proceedings of a Workshop—in Brief was reviewed by Robin Feldman, The University of California College of the Law, San Francisco; Peter Lurie, Center for Science in the Public Interest; and Joshua Sharfstein, Johns Hopkins University. Marilyn Baker, National Academies of Sciences, Engineering, and Medicine, served as the review coordinator.
COMMITTEE Barbara E. Bierer (Co-chair), Harvard Medical School and Brigham and Women’s Hospital; William B. Schultz (Co-chair), Zuckerman Spaeder; Halima Amjad, Johns Hopkins University School of Medicine; Rogério Gaspar, World Health Organization; William Hubbard, Independent Consultant; Frank W. Rockhold, Duke University Medical Center.
NATIONAL ACADEMIES OF SCIENCES, ENGINEERING, AND MEDICINE STAFF Steven Kendall, Senior Program Officer; Anne-Marie Mazza, Senior Director; Matt Cowan, Christine Mirzayan Science and Technology Policy Graduate Fellow (until May 2023).
SPONSORS This project was funded by Arnold Ventures, The Commonwealth Fund, and Kaiser Permanente.
The Commonwealth Fund is a national, private foundation based in New York City that supports independent research on health care issues and makes grants to improve health care practice and policy. The views presented here are those of the authors and not necessarily those of the Commonwealth Fund, its directors, officers, or staff.
SUGGESTED CITATION National Academies of Sciences, Engineering, and Medicine. 2023. The Food and Drug Administration’s Accelerated Approval Process for New Pharmaceuticals: Proceedings of a Workshop—in Brief. Washington, DC: The National Academies Press. https://doi.org/10.17226/27103.














