
Page 27
2—
Hormonally Active Agents
The term hormonally active agents (HAAs) is used in this report to describe substances that possess hormone-like activity, regardless of mechanism. Compounds with estrogenic activity received most of the initial concern regarding endocrine disruptors and are the most prominently studied HAAs. Therefore, they comprise the majority of the agents that could be critically evaluated by the committee. Antiestrogens, antiandrogens, aryl hydrocarbon (Ah) receptor agonists, and other toxicants acting through effects on hormonal systems are considered to a lesser extent but are no less important. Indeed, many published papers show that various hormonal systems (e.g., thyroid systems) are targets of HAAs. Thus, the emphasis of this chapter on xenoestrogens reflects the published literature, but does not imply any judgment about the relative importance of other HAAs.
This chapter identifies the synthetic HAAs that were evaluated by the committee and discusses naturally occurring HAAs, such as phytoestrogens. Although naturally occurring HAAs are not discussed extensively in the chapters on biologic effects, those compounds are ubiquitous in the environment and may confound background levels of exposure to HAAs (see Chapter 3).
One of the charges to the committee was to identify, if possible, the underlying mechanisms of action of HAA-related effects, and so this chapter also considers the mechanism of action of estrogenic compounds as a model of others wherein a ligand binds to a receptor and the resulting ligand receptor complex alters the transcription of mRNA, and ultimately cytoplasmic translation and protein synthesis. Thus, convincing evidence that an HAA can affect the endocrine system would be its ability to bind to classic hormone receptors and promote measurable responses, such as the induction of hormone-responsive genes or gene products. However, chemicals can disrupt hormonal processes, such ascontinue
Page 28
hormone synthesis, metabolism, organ-system interactions, and hypothalamicpituitary-gonadal-axis responses, by a variety of other mechanisms.
Hormone-Receptor-Mediated Actions
If an environmental factor influences a biologic organism, there must be physical interaction between the factor and the organism. Most physiologic systems involve detector or sensory molecules called receptors (Jensen and Jacobson 1960; Gorski et al. 1968). Receptors are present in the target cells of various organisms and interact with specific regulatory molecules present naturally, or in some cases, unnaturally, in the target cells' immediate environment. Many receptors-neural, hormonal, and developmental-are involved in different aspects of an organism's development and physiology.
The estrogen receptor (ER) is located in the cell nucleus and is a member of the superfamily of steroid and thyroid hormone receptors that act as ligand-induced nuclear transcription factors (Evans 1988). Steroid-thyroid hormone receptors contain several common structural domains that are conserved between the various members of this superfamily (Gronemeyer 1991; Truss and Beato 1993; Beato et al. 1995; Ing and O'Malley 1995; Mangelsdorf et al. 1995). Figure 2-1 is a model of ER ligands moving into a target cell and interacting with the ER in the nucleus. Steroid-hormone-binding domains are located in the Cterminus of the ER and are required for hormone-induced activation of nuclear transcription factors. Steroid-hormone-receptor-mediated induction of gene expression is a complex process that involves formation of the nuclear receptor complex, binding to hormone-responsive elements, and interaction with other transcription factors and coactivators associated with the RNA polymerase II transcription-initiation complex (Murdoch and Gorski 1991). The transactivation process is complex and requires interactions of proteins that bind to proximal and distal sequences in the 5'-promoter region of target genes. Other influences. including various coactivators, also modulate steroid-hormone-induced gene expression (Beato et al. 1995).
The nuclear localization of steroid-hormone receptors and the lipophilic nature of their ligands means that estrogen mimics or toxicants can readily access fundamental gene regulatory mechanisms in target cells. This group of regulators and their receptors constitute a system that is accessible to a variety of environmental factors that can readily disrupt normal physiologic mechanisms. The sections that follow discuss in more detail the nature of the ER and other steroid or steroidlike hormone receptors.
Estrogen Receptor
The ER is unusual because it recognizes a wide spectrum of chemicals as ligands and can form different architectural structures with them (Brzozowski etcontinue
Page 29
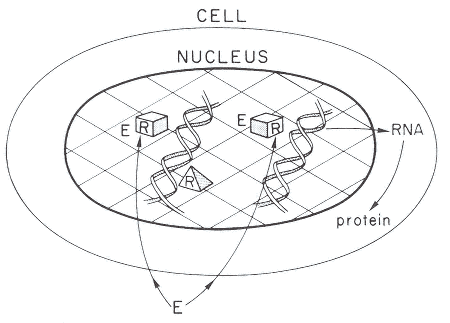
Figure 2-1
A simplified model of the cellular action of estrogens, androgens, thyroid hormone,
or many HAAs. Most HAAs are assumed to be able to diffuse through cell
membranes to reach the receptors located in the nucleus. R. receptor; pyramid,
unoccupied receptor; box, occupied receptor: E, environmental compound such
as estrogen, androgen, thyroid hormone, or HAA that can interact with the receptor.
Source: Gorski et al. 1986. Reprinted with permission from Recent Progress in
Hormone Research: copyright 1986, Academic Press, San Diego, Calif.
al. 1997). The shape of those structures affects the type of cellular and physiologic activities that can occur. For example, antiestrogenic versus estrogenic activities can occur because of the interactions of the ER with different ligands (Jordan and Murphy 1990). As is detailed in later chapters, many organic compounds have some estrogenic or antiestrogenic activity that can lead to a variety of biologic effects (see Chapters 5-10).
The diversity of ligands that interact with ER makes it difficult to predict structure-function relationships. Binding is extremely specific and capable of differentiating stereoisomers (Noteboom and Gorski 1965). Receptor affinity and occupancy are quantitative aspects of estrogen and antiestrogen binding to the ER that are important aspects of ER function and the effects of hormone mimics. Those quantitative aspects and the structures resulting from different ligand-receptor complexes are discussed below.break
Page 30
Receptor Affinity
The differences in dissociation rates of estrogens and antiestrogens from the ER have important biologic implications (Williams and Gorski 1972). The binding of estradiol to the ER has an equilibrium constant of ~ 0.1 nM because of a fast-forward rate of association and a relatively slow dissociation or off rate. At 0°C, the dissociation rate of estradiol is practically 0. At 37°C, the dissociation rate increases, resulting in a half-life of about 2 h (Kassis et al. 1986). Other ligands vary in their rates of dissociation; compounds such as estriol and estrone have rapid rates of dissociation and half-lives of just a few minutes (Pavlik and Katzenellenbogen 1980). There is little difference between the receptor affinity of human and other animal receptors, as would be expected because of the structural similarity of receptors in different species. It should be noted, however, that receptor half life is not the only determining factor relating the ligand to biologic activity. Systemic half life, which is effected by metabolism and clearance of the ligand, also must be considered when making assumptions with respect to biologic implications of the individual ligands.
Rapidly dissociating ligands, such as estriol, have been called "weak estrogens" (Stack and Gorski 1985). When they are injected in large quantities, these compounds often give a significant biologic response. However, only responses that occur shortly after the hormone is injected are observed. Cell replication and other, later, responses to estrogen are not observed. When the weak estrogens are injected at frequent intervals, changes in long-term responses also are detected (Stack and Gorski 1985). In cell culture, when weak estrogens are present at concentrations that cause a reasonable percentage of the receptors to be occupied, long- and short-term responses are observed (Stack and Gorski 1985). Thus. many compounds that can interact with the ER in a sustained manner can have a biologic effect even if they bind with a low-equilibrium affinity. However, in this case, the amount of such a compound must be great enough that it will occupy a significant number of estrogen-receptor-binding sites so as to induce an effect. A concentration 100 times that of estradiol would be necessary for a compound whose affinity is 1/100 that of estradiol. Many of the plant estrogens and environmental estrogens have affinities that are 1/100 to 1/1000 that of estradiol. Although ligands such as estradiol are classified as weak estrogens, that does not necessarily mean that they have fast clearance rates from the body. Dissociation rates and clearance are not directly related because multiple metabolic factors determine the rate of clearance. For example, clearance of a substance is related to plasma protein carrier kinetics, liver and kidney action, and metabolism of the agent within the target cell itself. In this regard, is it possible that estrogenic compounds with low receptor-binding affinities may still be biologically potent if they circulate for long periods within the body.
Accurately measuring rapidly dissociating estrogens is difficult when theycontinue
Page 31
are bound to the receptor. Under true equilibrium conditions, an accurate estimate of the affinity and the number of occupied receptors can be made. However, most rapid assays of receptor binding are not true equilibrium measurements. For example, in assays in which either the receptor or the ligand is bound to some solid support, such as charcoal, the bound material is no longer in equilibrium when the unbound ligand is washed away from the receptor. Thus, a low-affinity ligand—as are most of the environmental estrogens—is apt to dissociate from the receptor during the assay.
Receptor Occupancy
A rat uterine cell has about 20,000 ERs, approximately 10 nM. Clark et al. (1972) showed that under physiologic conditions, up to one-half of the receptors were occupied. Ruh et al. (1973) demonstrated that intact uteri that are incubated in a medium containing estradiol, estriol, or estrone exhibit various quantities of occupied ERs, which correlates with the induction of a specific protein in this tissue. MCF7 cells are derived from a human breast-cancer line and are widely used as a model cell-culture system of estrogen-regulated growth. MCF7 cells have about 50,000 ERs each. Maximal growth response occurs at about 0.1 nM estradiol, and a substantial response occurs at concentrations as low as 1 pM (Welshons and Jordan 1987). Under these cell culture conditions, we would expect that only 1,000 to 5,000 ERs per cell would be occupied. Thus, small pools of receptors that could be difficult to distinguish in a cell could be important biologically.
Diversity of Ligand-Receptor Complexes
There is evidence that estrogenic and antiestrogenic ligands cause differences in the structure of the ER (Hansen and Gorski 1985). When estradiol binds to the ER, the receptor's surface properties change significantly from hydrophobic to hydrophilic (Fritsch et al. 1992). The extent of this change in surface properties is different when 4-hydroxytamoxifen binds to the ER. Other estrogenic and antiestrogenic compounds could be studied to determine whether the structural characteristics of the bound ER are different when bound to estrogen mimics.
ER Subtypes
ER-mediated transactivation depends not only on cell- and gene-promotor context, but on ER subtype. Before 1996, most studies characterized estrogen responses of one ER (ERa); however, the recent discovery of an ER subtype (ERß) extends the potential tissue- and ligand-specific induction of estrogenic and antiestrogenic responses (Kuiper et al. 1996; Mosselman et al. 1996; Tremblay etcontinue
Page 32
al. 1997). ERa and ERß exhibit a common domain structure and high homology in the ligand-binding AF-2 and DNA-binding domains, but less than 25% homology in their N-terminal AF- 1 domains. ERa and ERß exhibit overlapping and differential expression in various tissues and cells, and both proteins readily form homo- or heterodimers that bind estrogen-responsive elements (EREs) in gel mobility shift assays (Cowley et al. 1997; Pace et al. 1997; Pettersson et al. 1997). ERa and ERß bind estrogenic steroid hormones, naturally occurring estrogenic compounds, and synthetic estrogens, including many of those presented in Tables 2-1 and 2-2 (Kuiper et al. 1997, 1998; Watanabe et al. 1997). A recent report described the relative binding affinities of 60 different estrogenic compounds to both ERa and ERß and only a few compounds exhibited major differences in binding to the ER-subtypes (Kuiper et al. 1998). For example, the relative binding affinity of genistein for ERß was greater than 20-fold higher than for ERa; however, in transactivation assays dependent on ERa or ERß, the activity of genistein was similar for both ER subtypes. Additional studies will be required to characterize fully ligand-induced ERa and ERß estrogenic and antiestrogenic activities.
Other Steroid or Steroid-like Hormone Receptors
Among the many nuclear receptors for steroid and related hormones are receptors for the androgens, thyroid hormones, and adrenal glucocorticoids, all of which have been implicated as targets of HAAs. The receptors have many characteristics in common with the ER. There is some controversy about whether the glucocorticoid receptor is nuclear or cytoplasmic when not bound to a ligand (Yamamoto et al. 1988). In either case, the receptor is always nuclear when bound to a glucocorticoid. The androgen receptor has a higher affinity for a testosterone metabolite, 5a-dihydrotestosterone (DHT), than it does for testosterone itself. DHT is formed in some but not all androgen target tissues; thus, the effects of testosterone in a whole organism will vary from tissue to tissue, depending on the presence of the 5a -reductase enzyme, which converts testosterone to DHT (Wilson et al. 1993). The ability of other compounds to react with the androgen receptor therefore depends on the nature of the compound and on whether it would behave more like testosterone or DHT or is a substrate for the reductase.
The thyroid hormone receptor is the protooncogene for the oncogene Erb-a (Sap et al. 1986). The mutated oncogenic form does not bind thyroid hormone but is constitutively activated. There are not as many studies that consider the range of compounds that can bind to the thyroid hormone receptor as there are for the ER, but the possibility of environmental sources of such compounds must be considered.break
Page 33
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(table continued on next page)break
Page 34
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(table continued on next page)break
Page 35
(table continued from previous page)break
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Page 36
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(table continued on next page)break
Page 37
(table continued from previous page)break
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Page 38
Hormonally Active Agents
Environmental Estrogens
Synthetic Estrogenic Compounds
Studies in the 1960s and 1970s characterized the estrogenic activity of several synthetic compounds or pesticides, including o,p'-DDT (dichlorodiphenyltrichloroethane), Kepone, phenolic compounds, PCB (polychlorinated biphenyl) mixtures, and some PCB congeners ( Tullner 1961; Bitman et al. 1968; Bitman and Cecil 1970; Ecobichon and MacKenzie 1974; Gellert 1978a,b; Hammond et al. 1979). PCB mixtures include planar/coplanar PCBs (i.e., PCBs with no ortho Cl substituents) and nonplanar PCBs (i.e., ortho-substituted PCBs). Hydroxy PCB congeners were later shown to bind to the ER and induce estrogenic responses in the female rodent uterus (Korach et al. 1988). With the development of the E-screen assay (Soto et al. 1995) and other in vitro bioassays, the list of synthetic estrogens has greatly expanded to include a spectrum of organochlorine compounds, phenolics, phthalates, and antioxidants (Soto et al. 1991, 1994, 1995: Krishnan et al. 1993; R. White et al. 1994; Jobling et al. 1995). Table 2-1 lists a representative group of synthetic compounds that have been shown to have estrogenic properties in a variety of assays. The structures of these compounds differ markedly with respect to molecular size and volume; substituent structure, and placement, thus demonstrating the unexpected and unusual diversity of ligands that bind to the ER (Figure 2-2). The relative potencies of synthetic estrogens is highly variable and depends on the target organ or cell and the specific end point. Bioavailability can be modified by pharmacokinetic variables and preferential binding to other cellular factors, such as steroid-hormone-binding globulin, transthyretin, and other proteins that interact with lipophilic molecules. Relative estrogenic potencies for several xenoestrogens have been determined in the E-screen cell proliferation assay (Soto et al. 1995), and the results are highly structure dependent. The potencies of several estrogenic organochlorine pesticides were approximately 106 (molar basis) times lower than that observed for estradiol. In contrast, 2',4',6'-trichloro-4-biphenylol and octylphenol were 10-4 and 3 x 10-4 times less active than estradiol in the MCF7 cell-proliferation assay. Studies that use an in vitro assay that incorporates changes in bioavailability associated with interactions with components of blood have shown that, for some environmental estrogens, in vivo potency (effective doses), particularly in fetuses, can be significantly different from previous estimates (vom Saal et al. 1995; Nagel et al. 1997).
Some HAAs have been detected in human tissues (adipose, serum, and milk) and in food (Kutz et al. 1991; Winter 1992), and there is potential for human exposures to many of these chemicals. For example, Brotons and co-workers (1995) detected bisphenol-A in liquids from food cans that contained a plastic-soft
Page 39
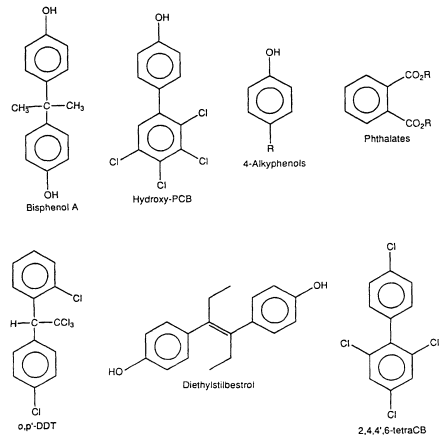
Figure 2-2
Some synthetic estrogens. The R values are limited relative to the potential estrogenic
activity of the molecules.
coated liner; bisphenol-A also was detected in saliva due to leaching from composites and sealants used in dentistry (Olea et al. 1996).
Natural Estrogenic Compounds
Several structural classes of naturally occurring compounds in plants as well as fungal metabolites exhibit estrogenic activity in various bioassays (Figure 2-3, Table 2-2). Flavonoids are present in fruits and vegetables, and high concentrations have been identified in various soy products. Humans consume approximately 1 g of flavonoids each day (Kuhnau 1976), and high consumption of these compounds has been correlated with lower incidence of stomach, colon, breast, and prostatecontinue
Page 40
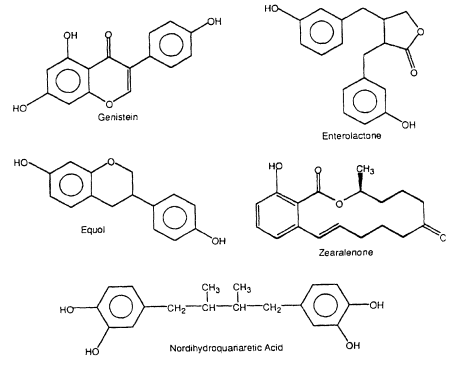
Figure 2-3
Naturally occurring estrogenic compounds.
cancers (Adlercreutz 1990). Two recent reviews summarize the health benefits of flavonoids (Bingham et al. 1998; Tham et al. 1998). Flavonoids elicit diverse ER-independent responses that could be important for their anticancer activity and that also could be responsible for estrogen-dependent adverse responses (Setchell and Adlercreutz 1988; Clarkson et al. 1995). However, it is apparent that, in addition to their estrogenic activity, flavonoids induce a broad spectrum of biologic responses that could contribute significantly to their anticarcinogenic activity (Adlercreutz et al. 1993a, 1995; Clarkson et al. 1995). Several studies characterized the estrogenic activity of flavonoids (Table 2-2); some of the compounds are weakly estrogenic in several assay systems. Flavonoid-derived compounds, such as coumestrol and equol; ligands, such as enterolactone and nordihydroguaiaretic acid (NDGA), which are present in food; and the fungal metabolites zearalenone, zearalenol, and zearalanol, also exhibit estrogenic activity.
As described above for synthetic estrogens, the potencies of naturally occurring estrogens are highly variable. For example, in the E-screen assay, zearalenol, zearalenone, and coumestrol were 100, 100, and 105 times less potent than was estradiol (Soto et al. 1995). In addition, the relative potency of phytoestrogenscontinue
Page 41
can be modulated in vitro via binding to serum proteins. For example, in the presence of serum from adult men, the uptake and binding of equol to estrogen receptors in MCF7 cells was 11 times higher than it was in a serum-free medium, suggesting that assays conducted in the absence of serum would underestimate the estrogenic potency of equol (Nagel et al. 1998).
Dietary intake of naturally occurring estrogens and their relative contribution of estrogen equivalents to the diet has been discussed (Kuhnau 1976; Verdeal and Ryan 1979). The potential mass balance of human exposure to natural and xenoestrogens should be further investigated. Although estrogenic flavonoids are rapidly metabolized, these compounds can be detected in serum and in urine (Adlercreutz et al. 1986, 1993b, 1995; Setchell et al. 1997).
Bioassays for Estrogenic Compounds
Several assay systems have been developed to measure the potential estrogenic activity of xenoestrogens and naturally occurring estrogenic compounds: Cell proliferation, ER binding, induction of estradiol-responsive genes or gene products, and transient or stably transfected cell-bioassay systems are summarized below. In Chapter 11, these assays are described in more detail, and their advantages and disadvantages are discussed.
The estrogenic effects of various chemicals depend on initial binding to the ER. Direct binding studies are impractical because the assays require a radio-ligand with high specific activity; however, competitive binding assays are routinely used to measure ER binding (Jordan et al. 1985; Miksicek 1995). These types of assays are conducted by determining the binding affinity of a test compound for the ER relative to a radiolabeled competitor with a known binding affinity (Gray et al. 1997).
The binding of steroid hormones to their respective receptors in the intact animal is complicated by biotransformation of some hormones into sulfates, glucuronides, and other oxidation products. In addition, steroid hormones can interact with blood constituents, such as the serum proteins, which can alter transport and cellular uptake of HAAs. For example, Welshons et al. (1997) and Nagel et al. (1998) have shown, using cell-proliferation assays and competitive-binding assays, that the presence of serum significantly affects the uptake and activity of estrogenic compounds, such as coumestrol, genistein, bisphenol, and octylphenol. The competition for plasma-carrier proteins sites, as well as for the receptor, is an equilibrium-competition process. The equilibrium of a steroid between the plasma proteins and other blood constituents and the plasma water and the cellular receptors dictates the amount of ligand receptor, which is assumed to be the biologically active form of the receptor (Montano et al. 1995).break
Page 42
The ability of estrogens to induce cellular proliferation in target organs is considered a hallmark of estrogen action (Hertz 1985). Therefore, a reliable bioassay for assessing estrogenicity would measure cell proliferation as an end point. This can be done in vitro by measuring mitotic indices in established cell lines derived from estrogen-responsive target organs. For example, Soto and Sonnenschein (1987, 1991) and Soto et al. (1994, 1995) have used the ''E-screen assay" to investigate the effects of various estrogenic compounds in the proliferation of MCF7 cells, a human breast-cancer cell line.
In vivo and in vitro studies have characterized several estrogen-induced genes or gene products that can be used as biologic markers of estrogen exposure: pS2, progesterone receptor, vitellogenin A2, cathepsin D, several protooncogenes, prolactin, transforming-growth factor a (TGFa), creatine kinase B, lactotransferrin, epidermal growth factor (EGF) receptor, calbindin D9k and D28k, heatshock protein 27, uterine peroxidase activity, insulinlike growth factor, binding protein-4, lactate dehydrogenase, and complement C3. Although each of these genes or gene products is induced by estrogenic compounds, the induction response could be specific to the target organ or cell, or gene products could be induced by other classes of HAAs. For example, prolactin synthesis might be induced by epidermal growth factor, thyrotropin-releasing factor, and phorbol esters (Ramsdell and Tashjian 1985). The synthesis of another estrogen-inducible marker, ovalbumin, is stimulated by other steroids, such as progesterone, and by glucocorticoids (Palmiter 1975). EGF induces several prototypical estrogenic responses in the mouse uterus and vagina (Nelson et al. 1991; Ignar-Trowbridge et al. 1992). The induction of estradiol-induced genes or gene products can be useful as a screening bioassay for estrogenic compounds; however, there is a potential for false positives because of overlapping inducibility by other hormones or chemicals.
Several in vitro bioassays that use recombinant receptor-reporter gene constructs have been developed to detect and quantitate estrogenic compounds and mixtures (Pons et al. 1990; Gagne et al. 1994; Jausons-Loffreda et al. 1994; Mäkelä et al. 1994; Jobling et al. 1995; Miksicek 1995; Ruh et al. 1995; Zacharewski et al. 1995). Transient transfection assays often use plasmids that contain 5'-flanking regions from estradiol-responsive genes, such as pS2 or vitellogenin A2, linked to reporter genes (chloramphenicol acetyl transferase, luciferase, ß-galactosidase). In some cell lines, estrogen responsiveness alsocontinue
Page 43
requires cotransfection of a human ER expression plasmid, and ER levels are controlled by varying the amount of cotransfected ER. Results using recombinant receptor-reporter genes in human cell lines indicate that their sensitivity is comparable to that of the E-screen assay. However, it should be noted that the E-screen assay measures the proliferative activity of a test chemical, which involves transcription of all genes required for cell growth, whereas other in vitro assays either measure ligand binding or induction of a single gene or gene product.
Estrogen-Receptor Antagonists
Estrogen-receptor antagonists, or antiestrogens, have been extensively investigated as drugs for treatment of ER-positive, hormone-responsive breast cancer (Lerner and Jordan 1990). Antiestrogens characteristically bind to the ER; however, their subsequent activities as ER antagonists or agonists depend on the animal species, target organ or cell, and response. Tamoxifen, a widely used nonsteroidal antiestrogen for treatment of breast cancer, has been extensively characterized as an ER agonist and as an ER antagonist (Jordan 1988; Lerner and Jordan 1990). Tamoxifen and its active metabolite, 4-hydroxytamoxifen, stimulate uterine growth and progesterone receptor expression in rodents, induce proliferation of MCF7 cells, and increase expression of several estrogen-responsive genes (Jordan and Prestwich 1978; Westley et al. 1984; Sonnenschein et al. 1985; Katzenellenbogen et al. 1987; May and Westley 1987; Thompson et al. 1989; Lerner and Jordan 1990). In contrast, the steroidal antiestrogens ICI 164,384 and ICI 182,780 exhibit primarily antiestrogenic activity (Thompson et al. 1989: Wakeling et al. 1991).
The possible antiestrogenic activity of synthetic estrogens and bioflavonoids have not been investigated extensively. Table 2-3 lists a few of the compounds that have been studied.
Mäkelä and co-workers (1994) report that coumestrol, genistein, and zearalenone—which are estrogenic—did not exhibit antiestrogenic activity in several cell-culture assays. In contrast, Markaverich and co-workers (1988) report that luteolin and quercetin inhibited estradiol-induced uterine hypertrophy in rats and estradiol-stimulated growth of MCF7 cells. Quercetin does not exhibit estrogenic activity, whereas luteolin is weakly estrogenic (i.e., shows partial agonist activity) in ER binding and in vitro reporter gene assays (Markaverich et al. 1995; Miksicek 1995). Ruh and co-workers (1995) report that the weakly estrogenic bioflavonoid naringenin inhibited estradiol-induced uterine hypertrophy, peroxidase activity, PR concentrations, and [3H]thymidine uptake in immature female rats and luciferase activity in MCF7 cells transiently transfected with the estrogen-responsive pS2-Luc plasmid. These data were obtained for individual bioflavonoids, and, coupled with a report suggesting that dietary soybeans are antiestrogenic (Mäkelä et al. 1995a), suggest that dietary compounds could be both estrogenic and antiestrogenic, depending on exposure.break
Page 44
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Page 45
Hydroxy-PCB congeners have been identified in human sera (Bergman et al. 1994). Those compounds have been synthesized (Safe et al. 1995) and their ER agonist-antagonist activities have been investigated in MCF7 cells and in estradiol-responsive, stably transfected HeLa cells (Moore et al. 1997). The seven major hydroxy-PCBs in human serum exhibited minimal to nondetectable competitive binding to the rat uterine ER and did not induce proliferation of MCF7 cells (10-5 to 10-8 M). The estrogenic activity of the hydroxy-PCB congeners was further investigated by two estrogen-responsive in vitro bioassays: In one, HeLa cells were stably transfected with a Gal4:human ER chimera and a 17merregulated luciferase reporter gene, and in the other, MCF7 cells were transiently transfected with a plasmid that contained an estradiol-responsive vitellogenin A2 promoter and a chloramphenicol acetyl transferase (CAT) reporter gene. None of the hydroxy-PCBs (10-5 to 10-8 M) significantly induced luciferase activity in the stably transfected HeLa cells or CAT activity in MCF7 cells. The antiestrogenic effects of the hydroxy-PCBs were investigated with the same bioassays in which the cells were treated with estradiol and with the hydroxy-PCBs. All of the hydroxy-PCB congeners inhibited one or more estrogenic response; one congener—2,2',3,4',5,5',6-heptachloro-4-biphenylol—inhibited estradiol-induced cell proliferation, CAT activity (MCF7 cells), and luciferase activity (HeLa cells). Pentachlorophenol also inhibits estradiol-induced trout ER and vitellogenin mRNA levels in trout hepatocytes, and it binds competitively to the ER (Flouriot et al. 1995). The estrogenic and antiestrogenic activities of other xenoestrogens should be investigated for multiple end points and for target organs and cells because estrogen and antiestrogenic responses could be highly specific.
Environmental Antiandrogens
Kelce and co-workers (1995) reported that p,p'-DDE (1, 1 -dichloro-2,2-bis(p-chlorophenyl)ethylene) competitively binds to the androgen receptor; however, in utero exposure of pregnant rats to 100 mg/kg/d (gestation d 14-18) resulted in retention of thoracic nipples, an antiandrogenic effect. Treatment of 25-d-old male rats (100 mg/kg/d until d 57) delayed the onset of puberty and, in castrated adult rats treated with testosterone to control for effects on testes, treatment with p,p'-DDE (200 mg/kg/d for 4 d) decreased seminal vesicle and prostate weight. Each response is consistent with antiandrogenic activity, and the results were confirmed in cell-culture studies using a recombinant chimeric androgen receptor and androgen-responsive promoter-reporter constructs. Because antiandrogens and estrogens can induce some of the same adverse responses (such as demasculinization) in male rodents, the hypothesized effects of xenoestrogens in male reproductive problems (Sharpe and Skakkebaek 1993) also should include "xenoantiandrogens." It is possible that other persistent organochlorine compounds are antiandrogens, and this should be further investigated. The identification of p,p'-DDE as an antiandrogen is important as well, because that compound is the majorcontinue
Page 46
persistent organochlorine pollutant in fish, wildlife, and human tissues; in places where DDT is still used as a pesticide, p,p'-DDE concentrations can be very high in human and wildlife tissues (Tanabe et al. 1994).
There also is evidence that the metabolites of vinclozolin, a dicarboximide fungicide, have antiandrogenic properties (Kelce et al. 1994). In vitro assays to determine the ability of vinclozolin to inhibit the conversion of testosterone to a more potent androgen (via 5 a-reductase) and to bind to the androgen receptor show that neither vincolzolin nor its degradation products (2-[[(3,5-dichlorophenyl)-carbamoyl]oxy]-2-methyl-3-butenoic acid and 3',5'-dichloro-2-hydroxy2-methylbut-3-enanilide) inhibit 5 a-reductase activity. Although the ability of vinclozolin to compete with androgen for binding to the androgen receptor is weak, its metabolites are effective androgen-receptor antagonists.
Aryl Hydrocarbon-Receptor Agonists
2.3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) and related halogenated aromatic hydrocarbons (HAHs) are industrial or combustion byproducts that have been extensively characterized as HAAs (Colborn et al. 1993; Peterson et al. 1993; Safe 1995). HAHs work through the Ah receptor-signaling pathway. Their biochemical and toxic responses in laboratory animals are variable and depend on the age, strain, sex, and species of the animal. In utero exposure to TCDD results in adverse effects in male and female rat offspring, and many of the responses observed in males are similar to those reported for antiandrogens and estrogens (Peterson et al. 1993). For example, male rats exposed to TCDD in utero exhibited decreased reproductive capacity, reduced androgenic status, and feminization of sexual behavior (Peterson et al. 1993). There also is concern regarding human offspring exposed to relatively large quantities of HAHs in utero (Peterson et al. 1993; Rogan 1995). An overall risk assessment of TCDD and related HAHs should consider exposures from synthetic-derived compounds and from naturally occurring Ah-receptor agonists, such as indole-3-carbinol, present in vegetables and other plant products (Wilker et al. 1996; Safe 1998).
TCDD and related Ah-receptor agonists also cause antiestrogenic activity in rodent uterus and mammary and human breast-cancer cell lines (Safe 1995). In those target tissues, TCDD inhibits estrogen-induced hypertrophy and growth of the rodent uterus and breast-cancer cells and mammary tumor growth in rodents. In addition, Ah-receptor agonists inhibit a spectrum of estrogen-induced genes and related activities. The mechanisms associated with the antiestrogenic activity involves crosstalk between the Ah- and estrogen-receptor-signaling pathways (Safe 1985). Bertazzi and co-workers (1993) report that women exposed to TCDD as a result of an industrial accident in Seveso, Italy, exhibited a decreased incidence of mammary and endometrial cancer, suggesting that comparable antiestrogenic responses were observed in cellular and animal models and humans. There also is evidence that other Ah-receptor agonists, such as polynuclear aro-soft
Page 47
matic hydrocarbons (PAHs) in tobacco smoke, induce antiestrogenic activity (Chaloupka et al. 1992). Two other studies (Lesko et al. 1985; Levi et al. 1987) report decreased incidence of endometrial cancer in tobacco smokers.
Other Hormonal Toxicants
Several other toxicants are hormone mimics or disrupt endocrine-response pathways. Hydroxy-PCBs and other hydroxylated organochlorine metabolites bind to transthyretin (Lans et al. 1993), and it has been suggested that these same compounds are thyroid-hormone-receptor agonists (Rickenbacher et al. 1986). A study of pregnant mice treated with 3,3',4,4'-tetrachlorobiphenyl showed significant accumulation of 3,3',4',5-tetrachloro-4-biphenylol bound to fetal transthyretin, accompanied by decreased fetal plasma T4 concentrations (Darerud et al. 1996). The transplacental effects of hydroxy-PCBs and other compounds that bind to transthyretin should be studied.
Mechanism of Estrogen Action
The ER is a ligand-induced transcription factor and is a member of the steroid/thyroid/retinoid nuclear-receptor superfamily (Mangelsdorf et al. 1995). The ER mediates temporal- and tissue-selective expression of specific genes, and these responses are dependent on ligand structure, cellular and gene-promoter context (Katzenellenbogen et al. 1996). The classical mechanism of ER action involves binding of the ligand-bound ER homodimer with perfect or imperfect palindromic 5'-promoter ERE motifs and subsequent interactions with the basal transcription factor complex. More recent studies have demonstrated that estrogen responsiveness is more complex and involves ER interactions with other nuclear factors, including coactivators and proteins such as p300/CBP that influence histone acetylation and chromatin structure (Mangelsdorf et al. 1995: Katzenellenbogen et al. 1996). The ER can also modulate gene expression by interacting with other DNA-bound transcription factors, and estrogen-mediated transcriptional activation can be observed through ER-API and ER- Spl interactions in which ER does not bind promoter DNA (Paech et al. 1997; Porter et al. 1997; Duan et al. 1998). The increasing complexity of ER action is consistent with the differential control of gene expression by ER and other members of the nuclear-receptor superfamily.
Even when it is not bound to an estrogen, the ER is found in the nucleus of its target cells. Although it is not clear what the ER is bound to in the nucleus, the complex chromatin structure of DNA and protein is a likely site. The chromatin of eukaryotic cells is arranged in an orderly manner, and different chromosomes have defined domains within the nucleus (Felsenfeld 1992). This is thought to be due to a network of proteins called the nuclear matrix, and there have been reports that ERs are associated with the nuclear matrix (Barrack 1987). ERs,continue
Page 48
with or without estrogen, also have a high affinity for specific DNA sequences, the EREs (Murdoch et al. 1990; Beato 1991). These sequences of 15-20 base pairs can convey estrogen responsiveness to reporter genes when ligated upstream of an appropriate promoter sequence. They have all the characteristics of enhancer sequences that are common regulatory elements in genes. Quantitative studies indicate that the ER, with or without ligand, has an affinity for its ERE of about 0.1 nM, which is similar to the affinity of the ER for estradiol or for diethylstilbestrol (DES) (Murdoch and Gorski 1991; Furlow et al. 1993). Thus, there are at least two high-affinity ligand sites for the steroid receptors, one for the steroid and one for DNA. This is illustrated in Figure 2-4, in which the ER is shown interacting with an ERE upstream from the site where transcription is initiated. The ER is also shown interacting with another protein at the ERE site. This could be another ER unit to yield a homodimer, or it could be another protein resulting in a heterodimer. Finally, the ER is shown interacting with another protein, which in turn is part of the transcription machinery or is closely associated with it.
Response elements vary in their sequence. The ER can distinguish single base-pair changes with dramatic changes in affinity of orders of magnitude. It also can recognize some DNA sequences that are quite different from the canonical vitellogenin gene ERE. It is undoubtedly the surface of the ERE that is recognized and not the sequence directly. Thus, there could be other response element sequences yet to be recognized.
When the unoccupied receptors are extracted from cells, they are found as large complexes with heat-shock proteins, proteins that are produced in response to environmental stress (Toft and Gorski 1966; Toft et al. 1967; Smith and Toftcontinue
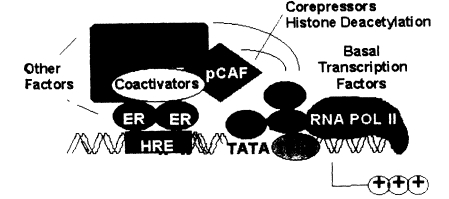
Figure 2-4
Transcriptional activation of estrogen-responsive genes: formation of ER-protein
complexes at distal responsive elements.
Page 49
1993). This is an area of considerable interest, and it is not clear how heat-shock proteins work in the functioning of the receptor. The heat-shock proteins could act as chaperons in the orderly folding of the receptor and translocation to the nucleus after its synthesis in the cytoplasm. It also has been suggested that the heat-shock proteins are present to prevent binding of the receptor to the DNA until the steroid binds to its receptor (Chambraud et al. 1990; Pratt et al. 1992).
After the estrogen or antiestrogen binds to the receptor in intact cells, the receptor ligand complex binds tightly to the nucleus and is difficult to extract. This complex is closely correlated quantitatively with the biologic response to estrogen. The binding of the ER to specific DNA sequences is part of the action of the ER but does not appear to be regulated by estrogens (Murdoch and Gorski 1991; Furlow et al. 1993). If the ER is bound to enhancerlike response elements without estrogen, the increase in affinity of the ER for the nucleus in intact cells exposed to estrogen must result from protein-protein interactions. The nature of these interactions has not been studied to any extent, but it is crucial for explaining how estrogenic compounds function.
There are published reports of the ER interacting with transcription factors associated with polymerase II, the RNA-transcribing enzyme known to synthesize mRNAs (Ing et al. 1992; Halachmi et al. 1994; Tsai and O'Malley 1994), as well as reports that corepressor proteins that interact with the transcription factors and with the ER are critical for the response to estrogens. This area is under active investigation, and more interactions have been reported than can be presented in a simple model. We can assume that, in the next few years, this area will be further elucidated so that a more detailed model of how estrogenic hormones activate transcription can be presented.
Another aspect of ER interactions with other proteins is that of self-interaction. Receptors are known to form homodimers, heterodimers, or both, at high concentrations in solution or when they bind to their respective response elements. In soluble systems, the ER forms homodimers that show cooperativity of estrogen binding (Notides et al. 1981; Sakai and Gorski 1984). However, there has been no evidence of cooperative estrogen binding or cooperative estrogen response in intact cells (Williams and Gorski 1974; Muller et al. 1985; Walent and Gorski 1990). This raises doubts about whether the ER is present as a homodimer in the intact cell. It could be that the ER pairs off with another protein to become a heterodimer that then binds to the ERE. Several other steroid receptors, such as the retinoic acid receptor, form heterodimers. This could be relevant to the problem of hormone mimics because the heterodimeric partner also might be influenced by environmental factors.
If ER interaction with nuclear proteins is the critical function of these receptors, then the possibility of variation in the nuclear composition of proteins that will interact with the ER becomes crucial for a cell's response to the ER and its ligands. Furthermore, if different ligands bound to the ER convey differentcontinue
Page 50
conformational states to the ER, then different ER-protein complexes could occur because of differences in the cell's nuclear protein composition and the estrogenic ligand.
Neural effects of estrogens could represent a more complex response to estrogen than that outlined above. Several reports indicate that estrogens regulate the neural system via changes in nuclear function similar to those described above. However, there are also reports in the literature of estrogen responses in neural cells that occur more rapidly than would be likely if they occurred as a result of changes in nuclear gene expression. Naftolin's group (Garcia-Segura et al. 1989, 1994) reported effects of 17ß-estradiol on neural membrane that occurred within 1 min of estrogen administration. Tamoxifen blocked the response, and 17a-estradiol had no effect. Pappas et al. (1994) showed that small numbers of ERs are found in cell membranes. Small pools of ERs could be missed by immunocytochemistry and other methods of ER detection currently in use.
Studies in animal models have shown that estrogens and androgens control epithelial cell numbers in their target organs by inhibiting cell death (Martin 1980), by inducing cell proliferation (Step I), and later by inhibiting cell proliferation (Step II) (Stormshak et al. 1976). Those effects can be segregated in experimental models by manipulating sex-hormone concentrations; this suggests that they are controlled by discrete mechanisms (Soto et al. 1986; Sonnenschein et al. 1994).
Three hypotheses postulate the role of estrogens on induction of cell proliferation (Step I):
· The direct positive hypothesis proposes that the estrogens themselves trigger the proliferation of their target cells (Stack and Gorski 1984).
· The indirect positive hypothesis proposes that estrogens induce the synthesis of growth factors that, in turn, cause proliferation of estrogen-sensitive cells via stroma-epithelium paracrine (Dickson et al. 1986) or autocrine (Cooke et al. 1986) interactions. However, growth factors administered to ER knockout mice failed to induce a proliferative effect in the female genital tract (Curtis et al 1996), which contradicts the indirect positive hypothesis.
· The indirect negative hypothesis posits that a plasma-borne inhibitory molecule (estrocolyone-I) inhibits the proliferation of estrogen-target cells (Soto and Sonnenschein 1985; Soto et al. 1992b; Sonnenschein et al. 1996). Estrogens could induce cell proliferation merely by neutralizing the effect of this serumborne inhibitor.
Continued treatment with estrogens results in a proliferative shutoff (Step II) of their target cells (Stormshak et al. 1976). Step II is considered a direct effect of estrogens in that it is ultimately mediated by hormones rather than by growth factors or inhibitors as postulated for Step I (Amara and Dannies 1983; Soto et al. 1986: Sonnenschein et al. 1994).break
Page 51
Modulation of Estrogen-Induced Responses
Estrogen-induced responses can be modulated by the internal cell environment. A complex of the antiestrogen 4-hydroxytamoxifen with the ER can have an agonistic effect in bone tissue; in breast tissue it is antiestrogenic (Jordan and Murphy 1990). The same cell type at different stages of maturity or development can react differently to the same steroid-ER complex because of the difference in internal cell environment. In molecular terms, this probably means interactions between the receptor and different cellular proteins present in the target cells. It is possible that some compounds might force a conformation of the ER that leads to interactions with different proteins in the nucleus. As the physiologic state of cells changes in response to a variety of influences, the complement of proteins present in the cell nucleus undoubtedly changes, and the ER complex then functions in an environment with a new array of proteins and transcription factors. This is an area of great interest to investigators that will influence the direction of research on hormone mimics.
An important relationship of ERs with environmental estrogens is seen in the presence of ERs in the very early stages of development of most vertebrates (Greco et al. 1993). This was brought to the attention of the medical community in the 1970s with the observation that the offspring of mothers who had been treated with large amounts of DES during high-risk pregnancies suffered abnormalities in various parts of male and female reproductive tracts. Most notable was the occurrence of vaginal adenocarcinomas, normally quite rare, that appeared after sexual maturity among some of the female offspring of DES-treated women. Although no similar cancers were observed in the treated mothers, daughters showed the response 15-20 yr after the mothers had received DES treatment. Thus, a combination of different cell environments and the presence of ERs in the early embryo fostered a totally unexpected problem that emphasized the importance of cellular environment influences on the response to chemical regulators.
The observations of embryonic effects were studied more carefully by McLachlan and co-workers (1980) in mice and rats and are detailed in the Appendix. The studies also point out that the presence of receptors in embryonic and fetal tissues presents a mechanism for estrogenic regulators to influence the developing organism. The normal function of the embryonic receptors is, as yet, not fully understood, but studies involving administration of estrogen agonists and antagonists during development suggest a role for estrogen receptors in the development of the brain, reproductive organs, and other tissues, such as bone (vom Saal et al. 1992; Greco et al. 1993; Newbold 1995).
Immunocytochemistry has shown that ERs are present in mouse blastocysts (Hou et al. 1996). Sensitive polymerase chain reaction (PCR) techniques showed that ER mRNA is present in the unfertilized mouse oocyte, and through the 32-64 cell blastocyst (Hou and Gorski 1993; Wu et al. 1992). PCR methods also werecontinue
Page 52
used to show that ER mRNA is present in human oocytes (Wu et al. 1993). Somewhat later in development, ERs are present in male and female reproductive tracts at the indeterminate stage (12- to 15-d-old embryo) of reproductive-tract development (Greco et al. 1993). From 15 d of embryonic age to the neonate, ERs increase in females and decrease in males, although substantial numbers remain even in males (Greco et al. 1993).
Immunocytochemistry techniques revealed that embryonic ERs are present in all cells of the blastocyst (Hou et al. 1996). However, during mouse development, the presence of the ER in various cell types changes. ERs are present in the gonads of males and in the reproductive tracts of males and females throughout fetal development and after birth (Greco et al. 1993). Although quantitative changes do occur; qualitatively, both sexes have estrogen-response systems that can respond to estrogens from physiologic or environmental sources.
Only one case of an ER-deficient human has been authenticated (Smith et al. 1994). This human male showed definite signs of estrogen insufficiency, but the observations raise the question of why no other ER-deficient humans or other animals have been reported. One suggestion is that the ER normally has an early embryonic role and that a deficiency of ER is lethal to the embryo. However, a line of transgenic knockout mice that were engineered to have a defective ER gene survive to adulthood and have normal gross external phenotypes (Lubahn et al. 1993; Korach et al. 1996). Although both sexes are infertile and have defects of the gonads indicative of low-estrogen responsiveness, these findings support the conclusion that prenatal development of the reproductive tract of both sexes appears to be independent of an ER-mediated response.
It is not clear what the prepuberal appearance of ERs signifies or whether they have any physiologic function. It is apparent that embryonic ERs can damage the organism when estrogens or potential estrogenic mimics present themselves in, the environment at sufficient concentrations.
Summary and Conclusions
The estrogen receptor and other steroid or steroidlike hormone receptors can bind with a range of compounds and as such are potential vehicles for HAAs to act upon and influence normal cellular pathways. Steroid hormone receptors control fundamental gene-regulatory mechanisms, and interaction of HAAs with these receptors may disrupt these processes. The potential for a biologic effect is dependent upon the cell type, concentration of an HAA in the target cell, and the binding affinity of that compound for a receptor. Most HAAs exhibit relatively low binding affinities for the ER, suggesting that relatively high concentrations of the compounds are required to induce a response. Some work suggests that HAAs may have a significant effect during cell growth and embryonic development, when organisms may be more sensitive to low concentrations of estrogens. HAAs, such as the partial agonist tamoxifen, may have different effects in differ-soft
Page 53
ent tissues and species. However, most of the compounds that exhibit estrogenic activity appear to elicit similar cellular responses when experiments account for differences in measured response times and differences in rates of dissociation of the compounds from the ER.
The estrogenic properties and potencies of several HAAs discussed in this chapter are highly variable and structure dependent. It should be emphasized that although in vitro assays are important screening tools for determining estrogenic activity, the results cannot be used to state with certainty that a particular HAA will interfere with normal hormonal pathways in an organism (see Chapter 11).
The multiple mechanisms of ER-mediated responses include interaction of the ER with specific DNA sequences, such as estrogen-responsive elements, interaction with other transcription factors, crosstalk with other signaling pathways, and cell-membrane-mediated responses. Estrogen and HAAs appear to induce estrogen-responsive cell proliferation and related responses through common pathways. Although both males and females are responsive to estrogens, ER expression and estrogen responsiveness of various cell types can change between prenatal and postnatal life. The role of HAAs on hormonally regulated cellular pathways is further discussed in the other chapters of this report.break



























