
3
The Electrometallurgical Process at Argonne National Laboratory
INTRODUCTION
The electrometallurgical process, as utilized in ANL’s demonstration project, consisted of a series of distinct steps to process SNF. As the demonstration project progressed, the EMT process was streamlined to generate three product streams. The details of the equipment used in the EMT process, and the waste forms and products that result from this process, are the focus of this chapter.
Figure 3.1 shows the electrometallurgical processing equipment associated with each of the EMT unit operations. The electrometallurgical treatment process for driver and blanket fuel comprises four stages: disassembly of the fuel elements with an element chopper; electrochemical removal of the uranium from the stainless-steel-clad fuel element pieces in an electrorefiner; consolidation of the electrodeposited uranium and removal of the entrained salt in a cathode processor; and finally, casting of the uranium product from the cathode processor into an ingot in a casting furnace. Down blending of the highly enriched uranium (HEU) driver fuel with depleted uranium takes place in both the cathode processor and the casting furnace. For blanket fuel, which is mostly depleted uranium, down blending is not necessary.
Two different electrorefiners are used to treat the segmented fuel elements. The Mark-IV electrorefiner employs a relatively simple mechanical design and has sufficient throughput to treat the relatively small amount of sodium-bonded driver fuel in the ANL-W inventory (3.5 metric tons of heavy metal), whereas the more sophisticated Mark-V electrorefiner with a much higher throughput is designed to treat the more plentiful sodium-bonded blanket fuel (56.5 metric tons). A detailed description of these electrorefiners as well as the equipment and procedures employed at each stage of the EMT treatment process is given below.
Fuel Element Choppers
The driver fuel assembly (Figure 3.2) consists of a hexagonal stainless-steel tube (cladding) containing 61 fuel elements loaded with HEU fuel.1 This fuel is a uranium alloy with 10 wt % Zr. The uranium consists of 63% atomic 235U after burnup. The gaps between the fuel and the inside walls of the cladding are filled with sodium to
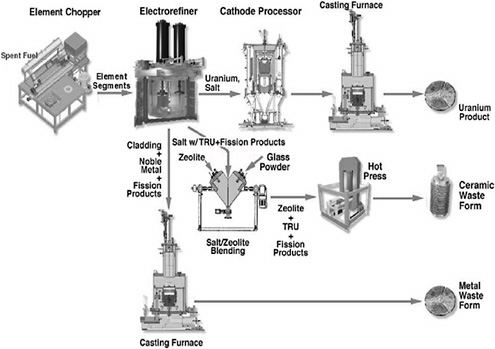
FIGURE 3.1 Equipment used in the treatment of EBR-II spent nuclear fuel.
SOURCE: Argonne National Laboratory.
provide a thermal bond; sufficient sodium-metal is also added to cover the top of the fuel. An argon-gas-filled expansion region extends from the top of the fuel/sodium to the top of each fuel element. An EBR-II driver assembly typically contains 4.5 kg of uranium.
The EBR-II blanket fuel is depleted uranium in a stainless-steel cladding (Figure 3.3). Each assembly contains 19 fuel elements; each element contains 5 slugs of uranium, each weighing 0.50 kg, for a total of 2.5 kg of uranium per element.2 The total weight of uranium in a blanket assembly is normally 47.5 kg.3 As is the case for the driver fuel, the gap between the fuel and the cladding is filled with sodium. The average discharge burnup for this fuel was approximately 1.2 wt %.
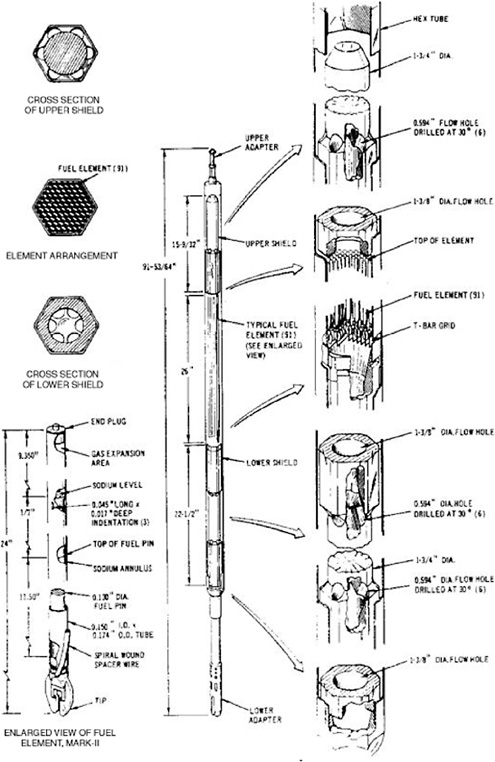
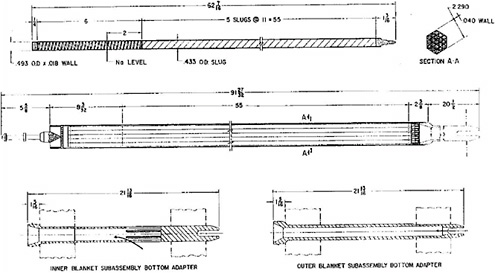
FIGURE 3.3 EBR-II inner and outer blanket fuel assemblies.
SOURCE: Argonne National Laboratory.
The fuel element choppers are pneumatic punch presses that have been modified with blades for shearing driver and blanket fuel elements into segments for loading into the anode compartments of the Mark-IV and Mark-V electrorefiners. The element choppers used for driver and blanket fuel differ somewhat in design but perform essentially the same function. The driver element chopper is programmed to chop that portion of the fuel element containing the fuel and sodium bond into pieces for loading into the fuel dissolution basket of the Mark-IV electrorefiner. The chopped blanket fuel segments are fed directly into the anode basket assemblies of the Mark-V anode-cathode module. As part of ANL’s demonstration project criterion that required a blanket throughput rate of 150 kg per month sustained for one month,4 the blanket element chopper was used to process 3.5 blanket fuel assemblies or 66 blanket fuel elements for a total of 164.4 kg of uranium.
Mark-IV Electrorefiner
The Mark-IV electrorefiner (ER; Figure 3.4) is designed specifically for treating the EBR-II driver fuel. It consists of a 1.0 m diameter, 1.0 m deep 2.25 Cr-1 Mo steel vessel with a cover. Four ports (approximately 25.4 cm. diameter) exist in the top of the ER for anode and cathode assemblies. The vessel is normally filled to a depth of 33 to 35.6 cm with molten LiCl-KCl eutectic and to about 10 cm with molten cadmium metal, which forms a pool in the bottom of the electrorefiner vessel. The cell is operated at 500 °C. There are four 25.4-cm (10-in.) ports
|
4 |
The full success criteria for the demonstration project, along with the goals to meet them, are included in Chapter 6. |

FIGURE 3.4 Schematic of the Mark-IV electrorefiner.
SOURCE: Argonne National Laboratory.
in the Mark-IV ER cover to permit electrode assemblies to be inserted in the molten salt electrolyte. Two ports are used for anodes and two are used for cathodes.
The anode assembly used in the Mark-IV ER consists of four rectangular, perforated stainless-steel compartments, called fuel dissolution baskets, arranged in the shape of a cross (Figure 3.5). Each anode assembly (four baskets) can hold about 8 kg of uranium, which is roughly the amount of uranium found in two driver fuel assemblies. The cathode is simply a mild steel rod or mandrel. The stainless-steel mandrel is normally immersed in the molten salt to a depth of 23 cm during the electrorefining of uranium. Both the anode assembly and the cathode mandrel are rotated to provide convective mass transport when the cell is in operation.
Steel scrapers are mounted on the inside wall of the electrorefiner to control the growth of the dendritic electrodeposit. These scrapers are placed near each cathode opening to prevent the uranium deposit from exceeding 25.4 cm in diameter; otherwise, it would be impossible to remove the uranium deposit through the cathode port at the conclusion of an electrorefiner run. Bottom scrapers are also necessary; they prevent the dendritic uranium deposit from contacting the cadmium pool in the bottom of the electrorefiner. During operation, the scrapers dislodge some of the uranium deposit, which falls into the cadmium pool and dissolves. A small stirring propeller inserted through the ER cell cover into the cadmium pool provides agitation.
The Mark-IV ER’s overall anode batch size of 16 kg, achieved by using dual anode assemblies in parallel with a single serial cathode. The efficiency of the overall electrorefining operation is enhanced by using a second

FIGURE 3.5 Fuel dissolution baskets.
SOURCE: Argonne National Laboratory.
cathode inserted into the molten salt through the fourth port. This second cathode allows the product from a direct transport run to be harvested from the off-line mandrel while the other serves as the cathode during a second transport run.
Cadmium chloride is added to the molten salt in the ER at the beginning of each run to oxidize some of the uranium metal in the chopped fuel segments to U3+; U3+ must be present in the melt to promote initial deposition at the cathode. Additions of CdCl2 are needed periodically to maintain the U3+ concentration at or above this level. The cell is also fitted with a continuously operated cadmium vapor trap to reduce the accumulation of cadmium on the electrode components.
The Mark-IV electrorefiner can be operated in three modes: (1) direct transport, (2) anodic dissolution, and (3) deposition. In the direct transport, the uranium is electrochemically oxidized from the chopped fuel elements as U3+, transported through the molten salt bath by forced convection, and then reduced to the metal at the stainless-steel mandrel. In the anodic dissolution mode, the fuel dissolution baskets serve as the anode, and the cadmium pool serves as the cathode: the uranium is electro-chemically oxidized from the chopped fuel elements as U3+, transported through the melt by forced convection, and then reduced at the surface of the cadmium pool, where it dissolves in the cadmium. (Reduction of plutonium at the cadmium cathode did not occur because of the low concentration of plutonium in the salt and careful voltage regulation.) In the deposition mode, the cadmium pool serves as the anode, and the stainless-steel mandrel serves as the cathode. The uranium is oxidized from the
cadmium pool and deposited on the steel mandrel. Anodic dissolution and deposition may thus be regarded as a two-step process. In the deposition mode, any uranium dendrites or pieces that dislodge from the mandrel dissolve in the cadmium and are oxidized and redeposited on the mandrel. The direct transport and the deposition mode were both used in ANL’s electrometallurgical demonstration project.
As part of ANL’s demonstration project,5 the Mark-IV electrorefiner was used to treat twelve driver assemblies over a two-month period at an average rate of 24 kg of uranium per month compared to the target criterion of 16 kg (~4 driver assemblies) per month over a three-month period.6
Mark-V Electrorefiner
The processing capacity of the Mark-IV ER is inadequate to permit timely processing of blanket fuel, which exists in much larger quantities than driver fuel. Thus, the low processing capacity of the Mark-IV ER was the driving force for the development of the Mark-V system, which was designed for processing of EBR-II blanket fuel. The basic difference between these two electrorefiners is the design of the anodes and cathodes, which allowed significant increase and throughput.
The 25.4-cm (10-in.) ACMs used in the Mark-V ER are derived from the 20.3-cm (8-in.) prototype ACM that was first tested in the Mark-III ER at ANL-E and a much larger 63.5-cm (25-in.) prototype ACM under exploration at ANL-E for use in a high-throughput electrorefiner (HTER)7 with an anode batch size of 150 kg. However, the HTER has been beset with a number of technical problems such as cell shorting and rotor stalling due to the buildup of a dense uranium deposit between the anode and cathode surfaces of the HTER, and it requires additional development (see Chapter 5). ANL apparently intends to use an ER based on this HTER to process fuel other than that associated with the EBR-II reactor.
Figure 3.6 is schematic of the Mark-V ER in use in the Fuel Conditioning Facility (FCF) at ANL-W. The Mark-V electrorefiner vessel is identical in dimensions to the Mark-IV vessel and holds approximately 650 kg of LiCl-KCl eutectic. Like the Mark-IV system, the Mark-V ER vessel has a cover equipped with four 25.4-cm-diameter ports for the insertion of electrodes. However, the Mark-V ER contains four integrated, independently operated anode-cathode modules (ACMs) that are considerably more complex than the simple rotating anode baskets and stainless-steel mandrel cathodes used in the Mark-IV system. Each ACM consists of two rotating anode basket assemblies with a total of nine anode baskets and three stationary cathode tubes (outer, middle, and inner) arranged in a concentric, circular configuration (Figure 3.7). Attached to the bottom of each ACM is a product collector basket. As the uranium deposit accumulates on the cathode surfaces, a series of closely spaced beryllia scraper blades mounted on the leading edges of the anode baskets remove the uranium deposits, which fall into the removable screen buckets positioned under the ACMs. The optimum position of these scrapers (leading or trailing), the best spacing between the scrapers and the cathode surfaces, and the best material to use for the fabrication of these scrapers have been determined by extensive trial-and-error experimentation. Each ACM has a capacity of about 37 kg of uranium, and each has its own 600-ampere power supply. With ACMs installed in all four ports of the Mark-V ER, the total anode batch size approaches 150 kg. This batch size is considerably greater than the 16-kg batch size of the Mark-IV ER and is roughly equivalent to about three blanket fuel assemblies. However, only three ACMs were installed in the ER to meet the ANL-W demonstration goal of 150 kg uranium/ month for blanket fuel processing.
Before operation of the Mark-V ER can begin, the ACMs must be charged with fuel segments produced by the blanket element chopper. After the ACMs are placed into the electrorefiner, the anode baskets are rotated counter-clockwise at 10 to 20 rpm while the cathode tube assembly and product collector are held stationary. For the
|
5 |
The full success criteria for the demonstration project, along with the goals to meet them, are included in Chapter 6. |
|
6 |
R.D. Mariani, D. Vaden, B.R. Westphal, D.V. Laug, S.S. Cunningham, S.X. Li, T.A. Johnson, J.R. Krsul, and M.J. Lambregts, Process Description for Driver Fuel Treatment Operations, NT Technical Memorandum No. 111, Argonne National Laboratory, Argonne, IL, 1999. |
|
7 |
E.C. Gay, S.R. Sherman, J.L. Willit, and R.K. Ahluwalia, Development of the Electrorefining Process for Blanket Fuel, NT Technical Memorandum No. 114, Argonne National Laboratory, Argonne, IL, 1999, p. 10. |

FIGURE 3.6 Schematic of the Mark-V electrorefiner.
SOURCE: Argonne National Laboratory.
demonstration project, UCl3 was added to the melt at a concentration of 4 to 7 wt %. Adding UCl3 and maintaining a steady-state concentration of it serve the same purpose as that described above for the Mark-IV ER. Because the Mark-V ER does not utilize a cadmium pool, it operates only in the direct transport mode. Thus, electrorefining is initiated by imposing a current between the anode and cathode surfaces of an ACM; this results in the direct transport of uranium from the fuel segments in the anode basket assembly to the cathode surfaces of the ACM.
For smooth ACM operation, it is optimal if the uranium forms on each cathode surface as loosely adherent dendrites so that it can be removed easily by the beryllia scrapers. Unfortunately, in addition to dendrites, dense deposits of uranium invariably occur that cannot be removed by the scrapers. If allowed to develop beyond a
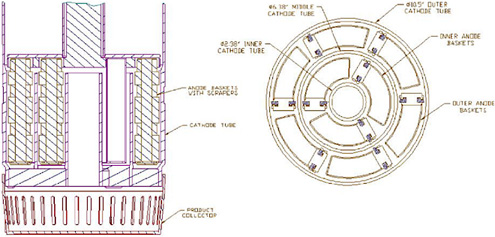
FIGURE 3.7 Anode-cathode configuration.
SOURCE: Argonne National Laboratory.
certain thickness, these dense uranium deposits can stall ACM rotation. It was determined that such deposits must be removed after about 200 ampere-hours of direct transport deposition to ensure continued successful operation of the Mark-V ER. These deposits are removed by electrochemical stripping, i.e., by reversing the direction of the current so that the cathode tubes become anodes and vice versa. A washing step is imposed between each deposition and stripping step. Washing consists of rotating the anode basket assembly in the ACM without the passage of current. An automated sequence involving a stripping step, washing step, deposition step, and a second washing step constituting an operational cycle appears to have mitigated the problem,8 with some decrease in overall efficiency.
The material that collects in the product collector basket consists of uranium and approximately 20 wt % salt. When the collector basket is removed from the electrorefiner, the uranium-salt mixture forms a dense mass that is tightly bound to the collector basket and cannot be removed by simply inverting the basket. Two methods were developed to remove the adherent product mass from the collector basket. The first utilizes a cutting/grinding tool, called the product-harvesting tool, to dig the solidified product out of the product collector basket. The second involves placing the product collector basket in a small oven, allowing the molten salt to drain away, and then inverting the product collector basket over an intermediate container. The material harvested from the product collector basket is sent to the cathode processor for further treatment.
Each ACM in the Mark-V ER is capable of processing about 87 to 100 kg of uranium per month. The use of additional ACMs does not necessarily lead to a proportional increase in the monthly process rate of uranium, because only one ACM can be serviced at a time. Therefore, anode basket and product collector changeover must be carefully coordinated with the start of each run to avoid conflicts that might arise from the need to service two ACMs simultaneously. As part of ANL’s demonstration project,9 the Mark-V ER processed the equivalent of 4.3 blanket assemblies (206 kg U per month), corresponding to a total uranium deposit mass of 204.9 kg. During this
|
8 |
This sequence is discussed in detail in Appendix B. |
|
9 |
The full success criteria for the demonstration project, along with the goals to meet them, are included in Chapter 6. |
time, 1.37 ACMs were active and the product collector rate was 212 g uranium/h per ACM. The average product collector fill rate per ACM limits the total processing rate. Each product collector can hold 13 kg of uranium product (uranium + 20 wt % salt), and three product collectors are required during the electrorefining of every 10 blanket elements.
Cathode Processor
The purpose of the cathode processor (Figure 3.8) is twofold: to remove entrained salt (and any cadmium) from the uranium electrodeposits by evaporation and to consolidate dendritic deposits. In the case of the driver fuel, depleted uranium must be added to the cathode processor to reduce the enrichment of the ER product to less than 50% for security reasons; this down blending is not necessary for blanket fuel (see Chapter 4). The cathode processor consists of an outer vessel; an induction-heated furnace assembly with coils, liner, and insulation; a crucible assembly with a graphite process crucible and cover, a radiation shield, a condenser, and a receiver crucible. A graphite furnace liner acts as the susceptor in the induction furnace, and the induction coils are passively cooled and protected by a vapor barrier. Entrained salt and any cadmium present in the driver fuel are removed from the molten uranium ER product by vacuum distillation and then deposited as a liquid in the receiver crucible. For convenience, the cathode processor is elevated above floor level so that the crucible assembly can be bottom loaded into the induction furnace. This position permits the process crucible to be loaded, emptied, and cleaned without affecting the furnace assembly.
The graphite process crucible used in the cathode processor is coated with a protective coating of ZrO2. Between 50 and 100 g of ZrO2 are applied, and the crucible is baked at 600 °C. The application of excess ZrO2 leads to the formation of dross and the subsequent loss of the uranium product due to the reaction
ZrO2 + U → UO2 + Zr.
The crucible must be cleaned and coated after each run. The use of a beryllia crucible is still under investigation to reduce the formation of dross.
The cathode processor sequence involves loading the crucible with product from the ER, positioning the crucible on the crucible assembly, and transporting the entire assembly into the cathode processor vessel. The cathode processor vessel is evacuated and heated in stages to a maximum temperature of ~1200 °C over a period of approximately 10 hours. ANL determined that operation of the cathode processor at temperatures above 1200 °C drastically increases dross formation and that holding the process crucible temperature slightly below the melting point of the uranium (mp = 1135 °C) for a short time improves the distillation of salt (and cadmium, if present). Thus, the crucible temperature is held at 1100 °C for one hour under vacuum before being increased to 1200 °C for one-half hour. After the process crucible has been held at 1200 °C for one-half hour, the current to the induction coils is discontinued, and the cathode processor is allowed to cool under vacuum to ambient temperature.
The final stage of the cathode processor operation involves lowering the process crucible assembly out of the furnace, placing it in a dumping fixture, and dislodging the resulting uranium ingot from the crucible. After several runs, the distillate containing salt and cadmium (Mark-IV) in the receiver crucible is carefully returned to the ER from which it originated so that any cadmium present in the distillate is not inadvertently added to the ER used to treat blanket fuel (Mark-V).
Casting Furnace
The casting furnace (Figure 3.9), which has evolved from earlier induction furnaces designed at ANL-E, provides a means to reduce the 235U enrichment of the driver fuel product from the cathode processor by the addition of depleted uranium resulting from the cathode processor and to consolidate further the uranium product. Its components are similar to those of the cathode processor except that there is no condenser stage and associated receiver crucible to collect the distillate. Like the cathode processor, the casting furnace is based on an induction furnace and a graphite crucible, and it can be evacuated. The furnace has two gas-tight flanges (top and bottom)


FIGURE 3.9 Schematic of the casting furnace.
SOURCE: Argonne National Laboratory.
that permit access to the casting crucible and other internal components. The crucible is normally loaded through the top flange employing the same fixture used to unload the cathode processor.
The operating parameters associated with the casting furnace include the crucible coating, temperature control, and pressure control. A crucible coating is required in the casting furnace to minimize the interaction of molten uranium with the graphite crucible and to prevent the cast ingot from adhering to the crucible walls. Yttria (Y2O3) was found to be suitable for this purpose and is applied in aerosol form. The loss of uranium due to reaction with the Y2O3 has been found by ANL to be much smaller than the loss due to reaction with the ZrO2 crucible lining in the cathode processor crucible.

FIGURE 3.10 Flow charts for the processing of driver and blanket fuel.
SOURCE: Argonne National Laboratory.
Figure 3.10 shows flow charts for the processing of blanket and driver fuel during the demonstration phase. The amounts of material that were treated as of July 1999 in the cathode processor and casting furnace are summarized in Appendix B.10 For the driver fuel, 468 kg of dendritic product (which consist of uranium and approximately 18% of the salt) was produced by the Mark-IV ER. This is the amount of uranium contained in roughly 100 driver assemblies.11 Likewise, the treatment of 5 of 25 blanket assemblies in the Mark-V ER produced 162.7 kg of electrorefined uranium (including approximately 18% salt).
FABRICATION OF WASTE FORMS
Metal Waste Form
Following the electrorefining operations the stainless-steel cladding hulls are left in the anode basket, along with the noble metal fission products (Zr, Mo, Ru, Rh, Pd, etc.), actinides and adhering salt electrolyte. The uranium content is about 4 wt %.

FIGURE 3.11 Iron-zirconium phase diagram.
SOURCE: Argonne National Laboratory.
Zr metal is added to improve performance properties of the final metal waste form and to produce a lower-melting point alloy. The target composition is stainless steel and 15 wt % Zr, with the allowable Zr concentration ranging from 5 to 20 wt %. As shown in the Fe-Zr phase diagram in Figure 3.11, an alloy of 13 wt % Zr is the low melting eutectic.
To distill the adhering processing salts, the material in the anode basket is placed in the cathode processor and heated to 1100 °C. The charge from the cathode processor is placed in an yttrium oxide crucible, is melted at approximately 1600 °C in the casting furnace in an Ar atmosphere, and then is cooled in the crucible or, on the research scale, cast into ingots. The actinides in the metal waste are primarily in this intermetallic phase. The ingot constitutes the metal waste form (MWF).
Ceramic Waste Form
Significant amounts of fission products and transuranic elements accumulate in the LiCl-KCl electrolyte used in the electrorefining process. Although the electrolyte can be recycled, its radioactive components must ultimately be disposed of as high-level radioactive waste (HLW). Vitrification methods used to immobilize other high-level radioactive waste materials cannot be used because glass cannot incorporate high concentrations of salt. For example, borosilicate glass waste forms can accommodate up to 20 mass % Na2O, but only about 1 mass % Cl. The ceramic waste form (CWF) has been developed to immobilize the active fission products (alkalis, alkaline earths, and rare earths) and transuranic elements of the electrolyte.
The ceramic waste form is produced in a batch process by mixing and blending the waste salt, periodically removed from the electrorefiner, with zeolite 4A at 500 °C to occlude the waste-loaded salt within the cages of the zeolite crystal lattice. The product of this step is called salt-loaded zeolite. Originally, the waste form was made from pure zeolite 4A, which is a fine (~5-mm average particle diameter) powder. Specimens made from this material are called “baseline” glass-bonded sodalite waste forms. In October 1997, a zeolite 4A material with particles nominally ranging from 74 to 250 μm in diameter (as defined by a specified sieve cut of −60+200 mesh) was selected for use in the CWF. It is supplied in a granular form and contains about ten mass percent of a proprietary clay binder. The granular zeolite 4A is much more easily handled; the waste forms made from this material are designated as “reference” glass-bonded sodalite. Salt-loaded zeolite is mixed with a borosilicate glass and consolidated at high temperature (850 to 900 °C) and pressure (14,500 to 25,000 psi) in a hot isostatic press (HIP) to make the final waste form. Under HIP conditions, the salt-loaded zeolite is converted to sodalite. The initial salt-loaded zeolite contained about 12 wt % occluded LiCl/KCl eutectic salt. The amount of occluded salt retained after the transformation in the sodalite phase is assumed to be the same as in the zeolite, an assumption that requires further experimental verification. The standard or reference ceramic waste form is made with 75 mass % salt-loaded zeolite and 25 mass % of a commercial glass that acts as a binder.
The fission product chlorides are allowed to build up in the salt until a sodium chloride limit is reached. The buildup of sodium chloride in the salt raises the melting point of the mixture. The sodium limit was established to provide a safe margin between the liquidus temperature of the salt and the operating temperature. For operation at 500 °C, the limit is estimated to be approximately 6 wt % sodium. Once the sodium limit is reached, enough salt is removed periodically from the electrorefiner to maintain the sodium concentration below the limit. This salt is disposed in the ceramic waste. During the demonstration phase the NaCl limit was not reached.
The steps for producing the ceramic waste form, as developed during ANL’s demonstration project, are depicted in Figure 3.12. The equipment shown in this schematic was used for operations with radioactive materials in the Hot Fuel Examination Facility (HFEF) at ANL-W. The demonstration-scale ceramic waste equipment was first tested out of cell with nonradioactive surrogates to develop operating parameters, because relatively little time was available for tests with radioactive materials before completing the demonstration.
A source of dried zeolite was needed for the ceramic waste form. The zeolite used is commercially available from Union Carbide Corporation/UOP, but it contains as much as 22 wt % moisture. A drying cycle was developed by ANL for 40 kg batches of zeolite. Thirty batches of zeolite were dried to less than 1 wt % using a 12-hour heating cycle.
The salt disposed of in the ceramic waste must be processed to an appropriate particle size for mixing with the zeolite. The salt first undergoes size reduction in a commercially available rock crusher. It is then fed into a Prater impact mill with a classifier where it is reduced to an average particle size of 245 μm. As part of nonradioactive testing, over 180 experiments were performed by ANL with the mill/classifier to evaluate the capabilities of the equipment and determine particle size requirements.
The salt and zeolite are combined and processed in a V-mixer where they are heated to 500 °C for approximately 15 h. In this operation the salt is occluded into the zeolite structure. Twenty-two V-mixer experiments were performed by ANL with nonradioactive materials. The V-mixer is also used to mix 25 wt % glass with the salt-loaded zeolite. The nominal batch size is 50 kg, but an 80-kg batch was also successfully tested by ANL.
The glass/salt-loaded zeolite mixture is loaded into stainless-steel cans for processing through the HIP. Before the cans are sealed, they are evacuated. A tungsten inert gas weld is used to seal the cans. More than 100 demonstration-scale HIP cans were processed prior to operations with radioactive materials. Two can styles, one of which is pictured in Figure 3.13, were successfully tested through the demonstration. The can was provided as part of a cooperative research and development agreement with the Australian Nuclear Science and Technology Organisation (ANSTO). The result of this extensive out-of-cell testing was the development of operating parameters and acceptance criteria for each of the ceramic waste form process steps.
Once all the equipment was installed in the HFEF, operations with radioactive materials commenced using the parameters developed from nonradioactive testing. The salt used for these tests came from the Mark-IV electrorefiner after treatment of 100 driver assemblies. The salt was removed from the electrorefiner using a tray system lowered into an electrorefiner port. After the tray filled with molten salt, it was removed, and the salt
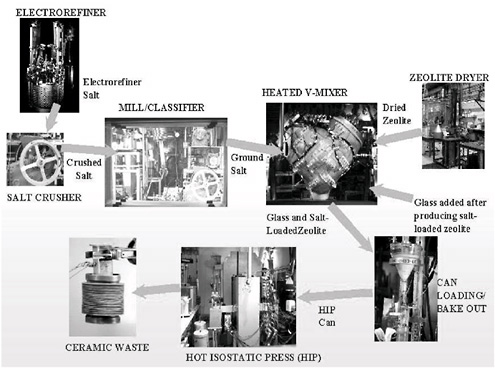
FIGURE 3.12 Ceramic waste process flow sheet.
SOURCE: Argonne National Laboratory.
solidified. The salt released easily from the tray. It was transferred by cask from the FCF to HFEF. It contained 9.4 wt % uranium chlorides, 0.5 wt % transuranic chlorides, and 5.2 wt % fission product chlorides. Four kilograms of salt were processed into a 50-kg batch of glass and salt-loaded zeolite. From this material, 10 HIP cans were processed through the HIP.
The cans used in the demonstration HIP are 4.5 in. in diameter (Figure 3.13). For inventory operations, the can diameter will be approximately 20 in. Scaling tests were performed using the nonradioactive materials. Three 8-in. diameter cans were first processed. The time at maximum temperature and the pressure in the HIP was increased from one hour for the demonstration-scale can to two hours for the 8-in. can. Characterization results indicated that the product in these cans had the same density, same phases present, and same performance based on leach tests as at the demonstration scale. Based on these results, two 18-in. diameter cans were processed. Characterization of these cans is ongoing. Initial X-ray diffraction results show that the same material phases are present in these cans as in cans of the other two sizes even though the hold time was 10 h.
In summary, the operating parameters for the production of the ceramic waste form have been developed and applied successfully to operations with radioactive materials at the demonstration scale. All the equipment for

FIGURE 3.13 Demonstration-scale hot isostatic pressure waste form can.
SOURCE: Argonne National Laboratory.
demonstration operations, except for the HIP, are close to the size required for inventory operations. Based on scaling tests with nonradioactive materials, the feasibility to scale HIP operations to production size has also been successfully demonstrated.
Pressureless Sintering
ANL is also investigating alternatives to use of the HIP for ceramic waste form fabrication. In particular, a process of pressureless sintering, or consolidation, is being studied that may have certain advantages over the HIP process.
The preparation steps for pressureless sintering are essentially identical to those for the HIP process. Zeolite A, glass binder, and waste-loaded salt are mixed in the V-mixer within the hot cell. The blended powder is poured into graphite molds (“setters”) that are passed through a tunnel kiln at a fixed temperature and time. The processed waste form is then removed from the mold, allowing re-use of the mold, and transferred into a storage container until it is loaded into a canister for final disposal.
Studies are being conducted to establish optimal fabrication procedures and to determine whether pressureless sintering can produce a suitable waste form. The current reference processing conditions for the kiln are 850 °C for approximately 4 hours at 1 atmosphere pressure of argon gas. Initial studies indicate that a higher volume percentage (50 vol %) of inert glass binder is needed to fabricate a reproducible waste form. Preliminary analysis indicates that the waste form produced by pressureless sintering has a density equal to that of the waste form produced by the HIP process, within analytical uncertainties. The short-term leaching characteristics of both waste forms are also essentially identical, although the committee has previously noted that such short-term tests may not
be fully indicative of long-term release-rate performance of waste forms under expected repository conditions.12 Furthermore, the heterogeneous nature of the multiphase ceramic waste form mandates examination of the microstructure and phase composition of as-produced waste forms.
Pressureless sintering may provide advantages over the HIP process during fabrication. As noted in the committee’s ninth report, the use of high-pressure HIP devices in hot cells may present unresolved safety and sustained-operation issues.13 Compared to the HIP process, the pressureless sintering method may also give a safer and easier pathway to volumetric scale-up of waste form fabrication. Further demonstration is needed and is being pursued by ANL. Higher rates (mass/time) of fabrication may also be achievable by pressureless sintering for equivalent-size fabrication equipment, although whether waste form fabrication is a rate-limiting step in the overall electrometallurgical processing technique is not apparent.
Recommendation: Studies to compare the type, abundance, and radionuclide inventory of minor and trace phases between ceramic waste forms produced by pressureless sintering versus the HIP process should be given high priority in the post-demonstration phase.
COMMITTEE ANALYSIS OF ALTERNATIVE METHODS FOR TREATMENT OF DOE SNF
A variety of other methods, besides electrometallurgical technology, are either in use or have been proposed for the treatment of spent nuclear fuel. The committee has evaluated alternatives to electrometallurgical technology in two of its reports.14,15 The following summarizes the committee’s evaluations of these alternative technologies in light of technical advances in the development of electrometallurgical treatment of spent nuclear fuel.16
Direct Disposal
From a materials and design standpoint, overpack technologies appear to be technically sound. However, the long-term durability of the proposed overpack container has not been demonstrated or documented. Without such a demonstration of extended containment, the ability of the HIC disposal concept to meet the stated safety goals proposed by the National Research Council is unknown.17 At the present time, direct emplacement of EBR-II SNF is precluded by DOE policy concerning acceptance of RCRA-designated mixed waste (which contains both hazardous and radioactive waste). Because of the presence of both metallic uranium and sodium, EBR-II SNF is categorized as an RCRA hazardous waste that is potentially both pyrophoric and reactive.
Glass Material Oxidation and Dissolution System (GMODS)
The basic concept of GMODS is to add unprocessed SNF and a sacrificial oxide to a glass melter at about 1000 °C, where a lead-borate glass acts as a solvent, and PbO oxidizes SNF metal components in situ to metal oxides which dissolve in the glass, and metallic lead. The lead metal (with dissolved metals) separates from the molten glass and sinks to the bottom of the melter. The GMODS process would be an attractive general approach that could be employed if it were successfully developed. However, considering the time period and cost necessary for development of the GMODS relative to the level of maturity of the EMT process, GMODS does not appear to be a viable alternative for processing only EBR-II SNF unless the process would also be applied to other DOE SNFs and miscellaneous fissile materials.
Melt and Dilute
The melt and dilute process chops SNF elements and melts them at approximately 650-850 °C, and then dilutes them by addition of depleted uranium and iron. No experimental work relevant to the processing of EBR-II SNF has been carried out. No development schedule or cost estimates were presented; therefore, the committee does not have sufficient information to evaluate melt and dilute as a viable alternative to EMT.
PUREX Process
The PUREX process is a counter-current solvent extraction method used to separate and purify uranium and plutonium from fission-product-containing SNF and irradiate uranium targets. The PUREX process is well developed and has been used to treat SNF and irradiated uranium for over 40 years. The development of a versatile head-end process to handle mechanical decladding, sodium removal, and zirconium sludge formation for EBR-II SNF for the SRS PUREX facility does not seem justified solely for the purpose of treating the relatively small quantity of EBR-II fuel that will remain after completion of the EMT demonstration. However, DOE would need to consider in the broader context whether developing a versatile head-end treatment step for the SRS PUREX facility is an attractive means for treating large quantities of DOE SNF (e.g., Zr or zircaloy-clad fuels such as N-reactor fuel) and other fissile-containing materials. A significant issue for treating EBR-II fuel at SRS by PUREX relates to public concerns about transportation of the fuel from the current storage site at ANL-W to SRS.18 A final consideration is that at the present time the PUREX canyons at SRS are due to be shut down and would not be available for this work according to Administration Policy.
Chloride Volatility
The unit operations for chloride volatility conist of: (1) a high-temperature chlorination step that operates at approximately 1500 °C and converts metallic fuel and cladding materials to gaseous chloride compounds, (2) a molten zinc chloride bed that removes the TRU chlorides and most of the fission products and operates at approximately 400 °C, (3) a series of fluidized beds and condensers operating at successively lower temperatures to condense zirconium tetrachloride, uranium hexachloride, and stannous tetrachloride, and (4) a zinc chloride regeneration/recycle process. The TRU and fission product chloride are then converted to either fluorides or oxides for final disposal. As proposed, this technology is applicable to a very limited range of fuel types. A reducing agent, such as carbon monoxide, would be needed in the chlorination step to prevent the formation of oxychloride compounds even for metallic fuel forms. The behavior of some of the constituents of EBR-II fuel, such as metallic sodium, may also limit the suitability of this process due to the accumulation of considerable amounts of sodium chloride during the chlorination step. Other unknown quantities, including the behavior of
stainless steel, in the chlorination step further decrease the potential applicability of this process to stainless steel-clad SNF (e.g., EBR-II fuel). At present, considering also cost and schedule, the chloride volatility process is not competitive with the current EMT process. In addition, the committee is aware that a significant amount of work on chloride volatility processes was conducted during the 1960s at ANL and ORNL.19 However, this earlier work does not suggest to the committee that this approach is an attractive alternative to EMT.
Plasma Arc
In this process, components of SNFs are melted and oxidized, with the help of an oxygen lance, in a rotating furnace containing molten ceramic materials at a temperature of 1600 °C or higher. Although plasma arc processing has been used successfully to treat nonradioactive and low-level radioactive wastes, significant research, development, and demonstration would be needed to process SNF because of the much higher fission product and fissile material content. With regard specifically to EBR-II fuels, extensive work would have to be performed to ensure that the plasma arc process would be compatible with safely processing potentially pyrophoric uranium, and volatile and reactive sodium metal. Unresolved safety issues at present preclude consideration of plasma arc processing as a viable alternative to the EMT process.




















