
Appendix C
PBPK Modeling
By R.S.H. Yang1
WHAT IS PHYSIOLOGICALLY BASED PHARMACOKINETICS? WHAT ARE THE DIFFERENCES BETWEEN PBPK AND CLASSICAL PHARMACOKINETICS?
Physiologically based pharmacokinetics (PBPK), as the name implies, is a special branch of pharmacokinetics where physiology and anatomy of the animal or human body, as well as the biochemistry of the chemical or chemicals of interest, are incorporated into the conceptual model for computer simulation. “Classical pharmacokinetics” refers to those empirical noncompartmental or compartmental pharmacokinetic studies routinely practiced in pharmaceutical industry (van de Waterbeemd and Gifford 2003). As will be illustrated later, the compartments of a PBPK model have anatomical and physiological significance. This is a major difference from empirical noncompartmental or compartmental pharmacokinetic modeling approaches. PBPK models can be used to describe concentration-time profiles in individual tissue/organ and in the plasma or blood. When the concentration of a certain target tissue, rather than the plasma concentration, is highly related to a compound’s efficacy or toxicity, PBPK modeling will be a more useful tool than classical pharmacokinetic models for describing PK/
PD relationship and thus make a better prediction of the time course of drug effects resulting from a certain dose regimen for the compound of interests. Furthermore, PBPK models in combination with absorption simulation and quantitative structure-activity relationship (QSAR) approaches will bring us closer to a full prediction of drug disposition for pharmaceutical new entities, and help streamline the selection of lead drug candidate in the drug discovery process (van de Waterbeemd and Gifford 2003). Lastly, unlike empirical noncompartmental and compartmental pharmacokinetics, PBPK modeling is a powerful tool for extrapolation, be it for inter-species, inter-routes, inter-doses, inter-life stages, etc.
The concepts of PBPK had its embryonic development in the 1920s and 1940s; for a more detailed early history, the readers are referred to a review (Yang et al. 2004) and a recently published book (Reddy et al. 2005). PBPK modeling blossomed and flourished in the late 1960s and early 1970s in the chemotherapeutic area mainly due to the efforts of investigators with expertise in chemical engineering process design and control. Two notable pioneers in this development are Dr. Kenneth B. Bischoff, then at University of Texas, Austin, TX, and Dr. Robert Dedrick of Biomedical Engineering and Instrumentation Branch, Division of Research Services, National Institute of Health, Bethesda, MD. Two timeless publications by these investigators are, respectively, “Drug Distribution in Mammals” (Bischoff and Brown, 1966) and “Animal Scale-Up” (Dedrick, 1973); these papers are highly recommended to those who are interested in PBPK modeling. In the mid 1980s, two papers on PBPK modeling of styrene and methylene chloride (Ramsey and Andersen 1984; Andersen et al. 1987) started yet another “revolution” in the toxicology and risk assessment arena. Today, there are more than 700 publications directly related to PBPK modeling on industrial chemicals, drugs, environmental pollutants, and simple and complex chemical mixtures (Reddy et al. 2005).
A PBPK model, graphically illustrated in Figure C-1, reflects the incorporation of basic physiology and anatomy. The compartments actually correspond to anatomic entities such as liver, lung, …etc., and the blood circulation conforms to the basic mammalian physiology. In this specific model, an actual published example on methylene chloride, it is quite obvious that the exposure route of interest is inhalation because the lung and gas exchange compartments are prominently displayed with intake (CI) and exhalation (CX) vapor concentrations indicated. Oral and/or dermal exposures may be added easily to the GI tract compartment or general venous circulation, respectively. Some tissues (e.g., richly (poorly) or slowly (rapidly) perfused tissues in Figure C-1) are “lumped” together because there is insufficient evidence to conclude that the component tissues in each of these compartment are kinetically distinct enough, for the specific chemical, to warrant individual separate compartments.
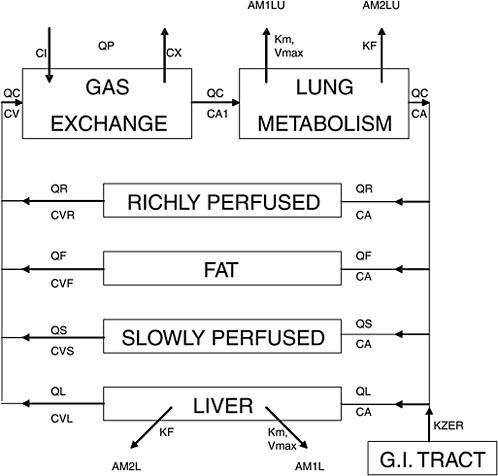
FIGURE C-1 A graphical representation of a physiologically based pharmacokinetic (PBPK) model for methylene chloride. Q represents blood flow; thus, QC, QP, QR, QF, QS, QL are cardiac output, pulmonary blood flow, and blood flows for richly perfused, fat, slowly perfused, and liver compartments, respectively. C represents concentration of the chemical we are studying; thus, CI, CX, CV, CA, CVR, CVF, CVS, CVL are concentrations in inhaled breath, exhaled breath, venous blood, arterial blood, richly perfused, fat, slowly perfused, and liver compartments, respectively. Km, Vmax, KF are metabolic parameters representing affinity constant, maximum rate constant for Michaelis-Menton enzyme kinetics (saturation or nonlinear kinetics), and first order rate constant (linear kinetics), respectively. AM1LU, AM2LU, AM1L, AM2L are, respectively, amount of metabolite 1 or 2 for lung (LU) and liver (L). Source: Andersen et al. 1987. Reprinted with permission; copyright 1987, Toxicology and Applied Pharmacology.
If one draws an analogy of the “scale-up” from a laboratory chemical engineering process to a chemical plant to the “scale-up” of a mouse to a human, one finds that both situations are governed by a great number of
physical and chemical processes. In mammals, the physical processes (i.e., mass balance, thermodynamics, transport, and flow) often vary in a predictable way. However, chemical processes such as metabolic reactions may vary greatly and less predictable among species. These physical and chemical processes interact in the body such that the pharmacokinetics of any given chemical between one species and another may be more (or less) predictable depending on the amount of background information available.
HOW DOES A PHYSIOLOGICALLY BASED PHARMACOKINETIC MODEL WORK?
A PBPK model applies fundamental physiological, biochemical, and engineering principles to describe the distribution and disposition of chemicals in the body at any given time. The process and approach may be summarized in a flow chart (Figure C-2). Once the chemical of interest and the problems needing to be addressed are identified, a thorough literature evaluation is conducted. Three sets of parameters are needed for PBPK model building: physiological parameters (e.g., ventilation rates, cardiac output, organs as % body weight), thermodynamic parameters (e.g., tissue partition coefficients, flow rates), and biochemical parameters (e.g., Km and Vmax). Most, if not all, of the parameters for laboratory animals are available in relevant literature such as the Biological Data Book, and the special report by the International Life Sciences Institute (ILSI) on the compilation of physiological parameters for PBPK models (Brown et al. 1997). When information gaps exist, the solution is either an empirical one via experimentation or through allometric extrapolation, usually based on a power function of the body weight (Lindstedt 1987).
The fundamentals of PBPK modeling are to identify the principal organs or tissues involved in the disposition of the chemical of interest and to correlate the chemical absorption, distribution, metabolism, and excretion within and among these organs and tissues in an integrated and biologically plausible manner. A scheme is usually formed where the normal physiology is followed in a graphical manner (i.e., a conceptual model as in Figure C-1). Within the boundary of the identified compartment (e.g., an organ or tissue or a group of organs or tissues), whatever ‘comes’ in must be accounted for via whatever ‘goes out’ or whatever is transformed into something else. This “mass balance” is expressed as a mathematical equation with appropriate parameters carrying biological significance. For instance, “a general equation, for any tissue or organ, is:
where Vi represents the volume of tissue group i, Qi is the blood flow rate to tissue group i, CAj is the concentration of chemical j in arterial blood, and Cij and CVijare the concentrations of chemical j in tissue group i and in the effluent venous blood from tissue i, respectively. Metabij is the rate of metabolism for chemical j in tissue group i; liver, being the principal organ for metabolism would have significant metabolism and, with some exception, usually Metabij is equal to zero in other tissue groups. Elimij represents the rate of elimination from tissue group i (e.g., biliary excretion from the liver), Absorpij represents uptake of the chemical from dosing (e.g., oral dosing), and PrBindingij represents protein binding of the chemical in the tissue.” All these terms are zero unless there is definitive knowledge that the particular organ and tissue of interest has such processes (Yang et al. 2005).
A series of such mass balance differential equations representing all of the interlinked compartments are formulated to express a mathematical representation, or model, of the biological system. This model can then be used for computer simulation to predict the time course behavior of any given parameter in the model.
For the most well-studied chemicals or drugs, it is likely that the biochemical constants such as Km’s and Vmax’s are known and readily retrievable from the information data base. However, it must be made clear here that the Km, Vmax, and KF (first order rate constant) in a PBPK model (known as in vivo Km, Vmax, KF for a given chemical) such as the ones given in Figure C-1 are hybrid constants of all the saturable or linear metabolic pathways, respectively, for the chemical of interest in the organ and/or body. They are different from the in vitro Km, Vmax, or KF of a given pure enzyme. While they are not directly interchangeable, the in vitro constants in the literature may be used to estimate in vivo constants for modeling purposes (Lipscomb et al. 1998; Kedderis and Lipscomb 2001). Also, for most well-known chemicals, it is likely that enough is known about the mechanism of toxicity to be incorporated into the model for computer simulation. The physiological constants such as organ volumes, blood flow rates, etc., are usually available in the literature for the common laboratory animals as well as humans. Therefore, at least in those instances of “well-known” chemicals, a model may be conceptually illustrated as in Figure C-1, and mathematically represented by a number of mass balance differential equations. Computer simulations may be made for any number of desired time-course endpoints such as the blood levels of the parent compound, liver level of a reactive metabolite, and similar information on different species, at lower or higher dose levels, and/or via a different route of exposure. The experimental pharmacokinetic data may then be compared with PBPK model simulation to see if it is superimposable upon each other. If this is indeed the case, the model is consistent with actual results. “Validation” of the PBPK model with data sets other than the working set
(or training set) to develop the model is necessary. We put the word “validation” in quotation mark because there are scientists who don’t really believe that there is such a thing as “model validation.” The most important thing to remember is that model is usually an over-simplification of reality; thus, “all models are wrong, some are useful” as stated by George Box. In such “validation” processes, the more the PBPK model is validated with other data sets, the more robust the model is in its predictive capability. Once “validated,” the PBPK model is ready for extrapolation to other animal species including humans. However, if the experimental data and PBPK model simulation are not consistent, the model might be deficient because critical scientific information might be missing or certain assumptions are incorrect. The investigator, with knowledge of the chemical and a general understanding of the physiology and biochemistry of the animal species, can design and conduct critical experiments for refining the model to reach consistency with experimentation (Figure C-2). This refinement
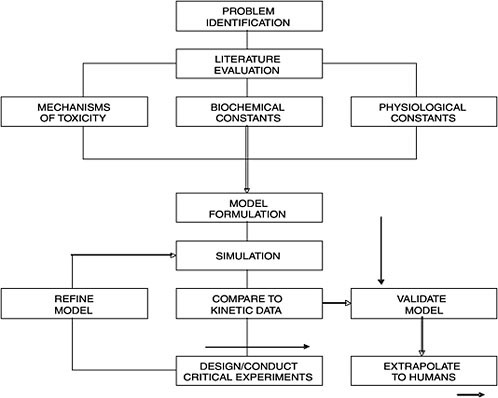
FIGURE C-2 A flow chart illustrating processes involved in physiologically based pharmacokinetics. Source: Andersen 1987. Reprinted with permission; copyright 1987, Toxicology and Applied Pharmacology.
process may be repeated again and again when necessary; such an iterative process is critically important for the development of a PBPK model. In that sense, PBPK modeling is a very good hypothesis-testing tool in toxicology and it may be utilized to conduct many different kinds of experiments on computer (i.e., in silico toxicology). It should be noted that there is always the possibility that a good model may not be obtained at the time because of the limitation of our knowledge on the chemical.
Physiologically Based Pharmacodynamic Modeling
Using plain English, pharmacokinetics can be considered as “What the body does to the chemicals,” and pharmacodynamics can thus be considered “What the chemicals do to the body.” Physiologically based pharmacodynamic (PBPD) modeling is therefore computer simulation of pharmacological or toxicological effects of chemicals or drugs, based on the biology of chemical/drug-receptor interactions. From the point of a chemical or a drug enters into the body to the point of pharmacological or toxicological effect, it is a continuum of pharmacokinetics and pharmacodynamics. It is difficult to distinguish where pharmacokinetics end and pharmacodynamics start. Having said that, however, this Appendix is centered around PBPK modeling; PBPD modeling is outside of the scope of this appendix.
Data Requirements for PBPK Modeling
What are the specific data needed for building PBPK models? Obviously, well conducted in vivo pharmacokinetic data are essential and usually the more the data sets (e.g., different doses, routes, species), the better. In each study, time-course blood and tissue concentration data are essential. These time-course data should include at least the following tissues and organs: blood (or plasma if blood cell binding is not an issue), liver (organ of metabolism), kidney (representing well-perfused organs/tissues), muscle (representing slowly-perfused organs/tissues), and target organ(s)/tissue(s). We also need other PBPK-modeling specific information such as: (1) physiological constants, including body size, organ and tissue volumes, blood flow and ventilation rates; (2) biochemical constants, including the chemical-specific metabolic rate constants such as Vmax and Km, partition coefficients for tissues; and (3) mechanistic factors such as target tissues, metabolic pathways, and receptor interactions. Enzyme kinetic data, particularly human data, of at least the key metabolic processes will be important for the PBPK model. In vitro determination of tissue partition coefficients and enzyme kinetic data are relatively straightforward and inexpensive. With modern genetic engineering technologies, many human enzymes are available commercially. Thus, heretofore unavailable human
enzyme kinetic information for many of the environmentally important chemicals are within easy reach for many laboratories.
PBPK Models for Chemical Interactions (Interactive PBPK Models)
Human exposure to chemicals is rarely, if ever, to single chemicals. The most ideal and scientifically defensible data requirement for establishing an interactive PBPK model is that each component chemical in the mixture already has its respectively established PBPK model and that there are many pharmacokinetic data sets in laboratory animals as well as in humans available for each of these component chemicals. The interactive PBPK model is then built on the basis of known pharmacokinetic interactions. For instance, the component chemicals may inhibit each others’ biotransformation. The individual PBPK models may then be linked together at the liver compartment by introducing competitive inhibition (or other types of inhibition) terms in the mass balance differential equation. Even though PBPK modeling of chemical mixtures is necessary in the cumulative risk assessment, this area is very complex and it is still an emerging field. For a more thorough discussion, the readers are referred to additional references on PBPK modeling of chemical mixtures (Krishnan et al. 2002; Yang and Andersen 2005).
REFERENCES
Andersen, M.E. 1987. Tissue dosimetry in risk assessment, or what’s the problem here anyway? Pp. 8-23 in Pharmacokinetics in Risk Assessment. Drinking Water and Health, Vol. 8. Washington, DC: National Academy Press.
Andersen, M.E., H.J. Clewell III, M.L. Gargas, F.A. Smith, and R.H. Reitz. 1987. Physiologically-based pharmacokinetics and the risk assessment for methylene chloride. Toxicol. Appl. Pharmacol. 87(2):185-205.
Bischoff, K.B., and R.G. Brown. 1966. Drug distribution in mammals. Chem. Eng. Prog. Symp. Ser. 62(66):33-45.
Brown, R.P., M.D. Delp, S.L. Lindstedt, L.R. Rhomberg, and R.P. Beliles. 1997. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol. Ind. Health 13(4):407-484.
Dedrick, R.L. 1973. Animal scale-up. J. Pharmacokinet. Biopharm. 1(5):435-461.
Krishnan, K., S. Haddad, M. Beliveau, and R. Tardif. 2002. Physiological modeling and extrapolation of pharmacokinetic interactions from binary to more complex chemical mixtures. Environ. Health Perspect. 110(Suppl. 6):989-994.
Kedderis, G.L., and J.C. Lipscomb. 2001. Application of in vitro biotransformation data and pharmacokinetic modeling to risk assessment. Toxicol. Ind. Health 17(5-10):315-321.
Lindstedt, S.L. 1987. Allometry: Body size constraints in animal design. Pp. 65-79 in Pharmacokinetics in Risk Assessment. Drinking Water and Health, Vol. 8. Washington, DC: National Academy Press.
Lipscomb, J.C., J.W. Fisher, P.D. Confer, and J.Z. Byczkowski. 1998. In vitro to in vivo extrapolation for trichloroethylene metabolism in humans. Toxicol. Appl. Pharmacol. 152(2):376-387.
Ramsey, J.C., and M.E. Andersen. 1984. A physiologically based description of the inhalation pharmacokinetics of styrene in rats and humans. Toxicol. Appl. Pharmacol. 73(1):159-175.
Reddy, M.B., R.S.H. Yang, H.J. Clewell III, and M.E. Andersen. 2005. Physiologically Based Pharmacokinetic (PBPK) Modeling: Science and Applications. Hoboken, NJ: Wiley. 420 pp.
van de Waterbeemd, H., and E. Gifford. 2003. ADMET in silico modeling: Towards prediction paradise. Nat. Rev. Drug Discov. 2(3):192-204.
Yang, R.S.H., and M.E. Andersen. 2005. PBPK modeling of chemical mixtures. Pp. 349-373 in Physiologically Based Pharmacokinetic (PBPK) Modeling: Science and Applications, M.B. Reddy, R.S.H. Yang, H.J. Clewell III, and M.E. Andersen, eds. Hoboken, NJ: Wiley.
Yang, R.S.H., M.E. Andersen, J.E. Dennison, Y.C. Ou, K.H. Liao, and B. Reisfeld. 2004. Physiologically based pharmacokinetic and pharmacodynamic modeling. Pp. 391-405 in Mouse Models of Cancer, E.C. Holland, ed. Hoboken, NJ: Wiley.
Yang, R.S.H., A.N. Mayeno, K.H. Liao, K.F. Reardon, and B. Reisfield. 2005. Integration of PBPK and Reaction Network Modelling: Predictive Xenobiotic Metabolomics. ALTEX. 22(Special Issue 2):328-334.









