
Reference Guide on DNA Identification Evidence
David H. Kaye, M.A., J.D., is Distinguished Professor of Law, Weiss Family Scholar, and Graduate Faculty Member, Forensic Science Program, The Pennsylvania State University, University Park, and Regents’ Professor Emeritus, Arizona State University Sandra Day O’Connor College of Law and School of Life Sciences, Tempe.
George Sensabaugh, D.Crim., is Professor of Biomedical and Forensic Sciences, School of Public Health, University of California, Berkeley.
CONTENTS
B. A Brief History of DNA Evidence
II. Variation in Human DNA and Its Detection
A. What Are DNA, Chromosomes, and Genes?
B. What Are DNA Polymorphisms and How Are They Detected?
2. Sequence-specific probes and SNP chips
C. How Is DNA Extracted and Amplified?
D. How Is STR Profiling Done with Capillary Electrophoresis?
E. What Can Be Done to Validate a Genetic System for Identification?
F. What New Technologies Might Emerge?
1. Miniaturized “lab-on-a-chip” devices
4. What questions do the new technologies raise?
III. Sample Collection and Laboratory Performance
A. Sample Collection, Preservation, and Contamination
1. What forms of quality control and assurance should be followed?
2. How should samples be handled?
IV. Inference, Statistics, and Population Genetics in Human Nuclear DNA Testing
A. What Constitutes a Match or an Exclusion?
B. What Hypotheses Can Be Formulated About the Source?
C. Can the Match Be Attributed to Laboratory Error?
D. Could a Close Relative Be the Source?
E. Could an Unrelated Person Be the Source?
1. Estimating allele frequencies from samples
2. The product rule for a randomly mating population
3. The product rule for a structured population
F. Probabilities, Probative Value, and Prejudice
1. Frequencies and match probabilities
G. Verbal Expressions of Probative Value
1. “Rarity” or “strength” testimony
2. Source or uniqueness testimony
V. Special Issues in Human DNA Testing
D. Offender and Suspect Database Searches
2. Near-miss (familial) searching
3. All-pairs matching within a database to verify estimated random-match probabilities
Deoxyribonucleic acid, or DNA, is a molecule that encodes the genetic information in all living organisms. Its chemical structure was elucidated in 1954. More than 30 years later, samples of human DNA began to be used in the criminal justice system, primarily in cases of rape or murder. The evidence has been the subject of extensive scrutiny by lawyers, judges, and the scientific community. It is now admissible in all jurisdictions, but there are many types of forensic DNA analysis, and still more are being developed. Questions of admissibility arise as advancing methods of analysis and novel applications of established methods are introduced.1
This reference guide addresses technical issues that are important when considering the admissibility of and weight to be accorded analyses of DNA, and it identifies legal issues whose resolution requires scientific information. The goal is to present the essential background information and to provide a framework for resolving the possible disagreements among scientists or technicians who testify about the results and import of forensic DNA comparisons.
Section I provides a short history of DNA evidence and outlines the types of scientific expertise that go into the analysis of DNA samples.
Section II provides an overview of the scientific principles behind DNA typing. It describes the structure of DNA and how this molecule differs from person to person. These are basic facts of molecular biology. The section also defines the more important scientific terms and explains at a general level how DNA differences are detected. These are matters of analytical chemistry and laboratory procedure. Finally, the section indicates how it is shown that these differences permit individuals to be identified. This is accomplished with the methods of probability and statistics.
Section III considers issues of sample quantity and quality as well as laboratory performance. It outlines the types of information that a laboratory should produce to establish that it can analyze DNA reliably and that it has adhered to established laboratory protocols.
Section IV examines issues in the interpretation of laboratory results. To assist the courts in understanding the extent to which the results incriminate the defendant, it enumerates the hypotheses that need to be considered before concluding that the defendant is the source of the crime scene samples, and it explores the
1. For a discussion of other forensic identification techniques, see Paul C. Giannelli et al., Reference Guide on Forensic Identification Expertise, in this manual. See also David H. Kaye et al., The New Wigmore, A Treatise on Evidence: Expert Evidence (2d ed. 2011).
issues that arise in judging the strength of the evidence. It focuses on questions of statistics, probability, and population genetics.2
Section V describes special issues in human DNA testing for identification. These include the detection and interpretation of mixtures, Y-STR testing, mitochondrial DNA testing, and the evidentiary implications of DNA database searches of various kinds.
Finally, Section VI discusses the forensic analysis of nonhuman DNA. It identifies questions that can be useful in judging whether a new method or application of DNA science has the scientific merit and power claimed by the proponent of the evidence.
A glossary defines selected terms and acronyms encountered in genetics, molecular biology, and forensic DNA work.
B. A Brief History of DNA Evidence
“DNA evidence” refers to the results of chemical or physical tests that directly reveal differences in the structure of the DNA molecules found in organisms as diverse as bacteria, plants, and animals.3 The technology for establishing the identity of individuals became available to law enforcement agencies in the mid to late 1980s.4 The judicial reception of DNA evidence can be divided into at least five phases.5 The first phase was one of rapid acceptance. Initial praise for RFLP (restriction fragment length polymorphism) testing in homicide, rape, paternity, and other cases was effusive. Indeed, one judge proclaimed “DNA fingerprinting” to be “the single greatest advance in the ‘search for truth’…since the advent of cross-examination.”6 In this first wave of cases, expert testimony for the prosecution rarely was countered, and courts readily admitted DNA evidence.
In a second wave of cases, however, defendants pointed to problems at two levels—controlling the experimental conditions of the analysis and interpreting the results. Some scientists questioned certain features of the procedures for extracting and analyzing DNA employed in forensic laboratories, and it became apparent
2. For a broader discussion of statistics, see David H. Kaye & David A. Freedman, Reference Guide on Statistics, in this manual.
3. Differences in DNA also can be revealed by differences in the proteins that are made according to the “instructions” in a DNA molecule. Blood group factors, serum enzymes and proteins, and tissue types all reveal information about the DNA that codes for these chemical structures. Such immunogenetic testing predates the “direct” DNA testing that is the subject of this chapter. On the nature and admissibility of the “indirect” DNA testing, see, for example, David H. Kaye, The Double Helix and the Law of Evidence 5–19 (2010); 1 McCormick on Evidence § 205(B) (Kenneth Broun ed., 6th ed. 2006).
4. The first reported appellate opinion is Andrews v. State, 533 So. 2d 841 (Fla. Dist. Ct. App. 1988).
5. The description that follows is adapted from 1 McCormick on Evidence, supra note 3, § 205(B).
6. People v. Wesley, 533 N.Y.S.2d 643, 644 (Alb. County. Ct. 1988).
that declaring matches or nonmatches in the DNA variations being compared was not always trivial. Despite these concerns, most cases continued to find the DNA analyses to be generally accepted, and a number of states provided for admissibility of DNA tests by legislation. Concerted attacks by defense experts of impressive credentials, however, produced a few cases rejecting specific proffers on the ground that the testing was not sufficiently rigorous.7
A different attack on DNA profiling begun in cases during this period proved far more successful and led to a third wave of cases in which many courts held that estimates of the probability of a coincidentally matching DNA profile were inadmissible. These estimates relied on a simple population genetics model for the frequencies of DNA profiles, and some prominent scientists claimed that the applicability of the mathematical model had not been adequately verified. A heated debate on this point spilled over from courthouses to scientific journals and convinced the supreme courts of several states that general acceptance was lacking. A 1992 report of the National Academy of Sciences proposed a more “conservative” computational method as a compromise,8 and this seemed to undermine the claim of scientific acceptance of the less conservative procedure that was in general use.
In response to the population genetics criticism and the 1992 report came an outpouring of critiques of the report and new studies of the distribution of the DNA variations in many populations. Relying on the burgeoning literature, a second National Academy panel concluded in 1996 that the usual method of estimating frequencies in broad racial groups generally was sound, and it proposed improvements and additional procedures for estimating frequencies in subgroups within the major population groups.9 In the corresponding fourth phase of judicial scrutiny of DNA evidence, the courts almost invariably returned to the earlier view that the statistics associated with DNA profiling are generally accepted and scientifically valid.
In the fifth phase of the judicial evaluation of DNA evidence, results obtained with the newer “PCR-based methods” entered the courtroom. Once again, courts considered whether the methods rested on a solid scientific foundation and were generally accepted in the scientific community. The opinions are practically unanimous in holding that the PCR-based procedures satisfy these standards. Before long, forensic scientists settled on the use of one type of DNA variation (known as short tandem repeats, or STRs) to include or exclude individuals as the source of crime scene DNA.
7. Moreover, a minority of courts, perhaps concerned that DNA evidence might be conclusive in the minds of jurors, added a “third prong” to the general-acceptance standard of Frye v. United States, 293 F. 1013 (D.C. Cir. 1923). This augmented Frye test requires not only proof of the general acceptance of the ability of science to produce the type of results offered in court, but also of the proper application of an approved method on the particular occasion. For criticism of this approach, see David H. Kaye et al., supra note 1, § 6.3.3(a)(2).
8. National Research Council, DNA Technology in Forensic Science (1992) [hereinafter NRC I].
9. National Research Council, The Evaluation of Forensic DNA Evidence (1996) [hereinafter NRC II].
Throughout these phases, DNA tests also exonerated an increasing number of men who had been convicted of capital and other crimes, posing a challenge to traditional postconviction remedies and raising difficult questions of postconviction access to DNA samples.10 The value of DNA evidence in solving older crimes also prompted extensions of some statutes of limitations.11
In sum, in little more than a decade, forensic DNA typing made the transition from a novel set of methods for identification to a relatively mature and well-studied forensic technology. However, one should not lump all forms of DNA identification together. New techniques and applications continue to emerge, ranging from the use of new genetic systems and new analytical procedures to the typing of DNA from plants and animals. Before admitting such evidence, courts normally inquire into the biological principles and knowledge that would justify inferences from these new technologies or applications. As a result, this guide describes not only the predominant STR technology, but also newer analytical techniques that can be used for forensic DNA identification.
Human DNA identification can involve testimony about laboratory findings, about the statistical interpretation of those findings, and about the underlying principles of molecular biology. Consequently, expertise in several fields might be required to establish the admissibility of the evidence or to explain it adequately to the jury. The expert who is qualified to testify about laboratory techniques might not be qualified to testify about molecular biology, to make estimates of population frequencies, or to establish that an estimation procedure is valid.12
10. See, e.g., Osborne v. District Attorney’s Office for Third Judicial District, 129 S. Ct. 2308 (2009) (narrowly rejecting a convicted offender’s claim of a due process right to DNA testing at his expense, enforceable under 42 U.S.C. § 1983, to establish that he is probably innocent of the crime for which he was convicted after a fair trial, when (1) the convicted offender did not seek extensive DNA testing before trial even though it was available, (2) he had other opportunities to prove his innocence after a final conviction based on substantial evidence against him, (3) he had no new evidence of innocence (only the hope that more extensive DNA testing than that done before the trial would exonerate him), and (4) even a finding that he was not source of the DNA would not conclusively demonstrate his innocence); Skinner v. Switzer, 131 S. Ct. 1289 (2011); Brandon L. Garrett, Judging Innocence, 108 Colum. L. Rev. 55 (2008); Brandon L. Garrett, Claiming Innocence, 92 Minn. L. Rev. 1629 (2008).
11. See, e.g., Veronica Valdivieso, DNA Warrants: A Panacea for Old, Cold Rape Cases? 90 Geo. L.J. 1009 (2002).
12. Nonetheless, if previous cases establish that the testing and estimation procedures are legally acceptable, and if the computations are essentially mechanical, then highly specialized statistical expertise might not be essential. Reasonable estimates of DNA characteristics in major population groups can be obtained from standard references, and many quantitatively literate experts could use the appropriate formulae to compute the relevant profile frequencies or probabilities. NRC II, supra note 9, at 170. Limitations in the knowledge of a technician who applies a generally accepted statistical procedure can be explored on cross-examination. See Kaye et al., supra note 1, § 2.2. Accord Roberson v. State, 16 S.W.3d 156, 168 (Tex. Crim. App. 2000).
Trial judges ordinarily are accorded great discretion in evaluating the qualifications of a proposed expert witness, and the decisions depend on the background of each witness. Courts have noted the lack of familiarity of academic experts—who have done respected work in other fields—with the scientific literature on forensic DNA typing and on the extent to which their research or teaching lies in other areas.13 Although such concerns may affect the persuasiveness of particular testimony, they rarely result in exclusion on the grounds that the witness simply is not qualified as an expert.
The scientific and legal literature on the objections to DNA evidence is extensive. By studying the scientific publications, or perhaps by appointing a special master or expert adviser to assimilate this material, a court can ascertain where a party’s expert falls within the spectrum of scientific opinion. Furthermore, an expert appointed by the court under Federal Rule of Evidence 706 could testify about the scientific literature generally or even about the strengths or weaknesses of the particular arguments advanced by the parties.
Given the great diversity of forensic questions to which DNA testing might be applied, it is not feasible to list the specific scientific expertise appropriate to all applications. Assessing the value of DNA analyses of a novel application involving unfamiliar species can be especially challenging. If the technology is novel, expertise in molecular genetics or biotechnology might be necessary. If testing has been conducted on a particular organism or category of organisms, expertise in that area of biology may be called for. If a random-match probability has been presented, one might seek expertise in statistics as well as the population biology or population genetics that goes with the organism tested. Given the penetration of molecular technology into all areas of biological inquiry, it is likely that individuals can be found who know both the technology and the population biology of the organism in question. Finally, when samples come from crime scenes, the expertise and experience of forensic scientists can be crucial. Just as highly focused specialists may be unaware of aspects of an application outside their field of expertise, so too scientists who have not previously dealt with forensic samples can be unaware of case-specific factors that can confound the interpretation of test results.
II. Variation in Human DNA and Its Detection
DNA is a complex molecule that contains the “genetic code” of organisms as diverse as bacteria and humans. Although the DNA molecules in human cells are
13. E.g., State v. Copeland, 922 P.2d 1304, 1318 n.5 (Wash. 1996) (noting that defendant’s statistical expert “was also unfamiliar with publications in the area,” including studies by “a leading expert in the field” whom he thought was “a ‘guy in a lab somewhere’”).
largely identical from one individual to another, there are detectable variations—except for identical twins, every two human beings have some differences in the detailed structure of their DNA. This section describes the basic features of DNA and some ways in which it can be analyzed to detect these differences.
A. What Are DNA, Chromosomes, and Genes?
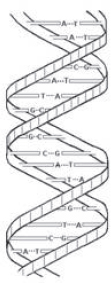
The DNA molecule is made of subunits that include four chemical structures known as nucleotide bases. The names of these bases (adenine, thymine, guanine, and cytosine) usually are abbreviated as A, T, G, and C. The physical structure of DNA is often described as a double helix because the molecule has two spiraling strands connected to each other by weak bonds between the nucleotide bases. As shown in Figure 1, A pairs only with T and G only with C. Thus, the order of the single bases on either strand reveals the order of the pairs from one end of the molecule to the other, and the DNA molecule could be said to be like a long sequence of As, Ts, Gs, and Cs.
Figure 1. Sketch of a small part of a double-stranded DNA molecule. Nucleotide bases are held together by weak bonds. A pairs with T; C pairs with G.
Most human DNA is tightly packed into structures known as chromosomes, which come in different sizes and are located in the nuclei of cells. The chromosomes are numbered (in descending order of size) 1 through 22, with the remaining chromosome being an X or a much smaller Y. If the bases are like letters, then each chromosome is like a book written in this four-letter alphabet, and the nucleus is like a bookshelf in the interior of the cell. All the cells in one
individual contain identical copies of the same collection of books. The sequence of the As, Ts, Gs, and Cs that constitutes the “text” of these books is referred to as the individual’s nuclear genome.
All told, the genome comprises more than three billion “letters” (As, Ts, Gs, and Cs). If these letters were printed in books, the resulting pile would be as high as the Washington Monument. About 99.9% of the genome is identical between any two individuals. This similarity is not really surprising—it accounts for the common features that make humans an identifiable species (and for features that we share with many other species as well). The remaining 0.1% is particular to an individual. This variation makes each person (other than identical twins) genetically unique. This small percentage may not sound like a lot, but it adds up to some three million sites for variation among individuals.
The process that gives rise to this variation among people starts with the production of special sex cells—sperm cells in males and egg cells in females. All the nucleated cells in the body other than sperm and egg cells contain two versions of each of the 23 chromosomes—two copies of chromosome 1, two copies of chromosome 2, and so on, for a total of 46 chromosomes. The X and Y chromosomes are the sex-determining chromosomes. Cells in females contain two X chromosomes, and cells in males contain one X and one Y chromosome. An egg cell, however, contains only 23 chromosomes—one chromosome 1, one chromosome 2,…, and one X chromosome—each selected at random from the woman’s full complement of 23 chromosome pairs. Thus, each egg carries half the genetic information present in the mother’s 23 chromosome pairs, and because the assortment of the chromosomes is random, each egg carries a different complement of genetic information. The same situation exists with sperm cells. Each sperm cell contains a single copy of each of the 23 chromosomes selected at random from a man’s 23 pairs, and each sperm differs in the assortment of the 23 chromosomes it carries. Fertilization of an egg by a sperm therefore restores the full number of 46 chromosomes, with the 46 chromosomes in the fertilized egg being a new combination of those in the mother and father. The process resembles taking two decks of cards (a male and a female deck) and shuffling a random half from the male deck into a random half from the female deck, to produce a new deck.
During pregnancy, the fertilized cell divides to form two cells, each of which has an identical copy of the 46 chromosomes. The two then divide to form four, the four form eight, and so on. As gestation proceeds, various cells specialize (“differentiate”) to form different tissues and organs. Although cell differentiation yields many different kinds of cells, the process of cell division results in each progeny cell having the same genomic complement as the cell that divided. Thus, each of the approximately 100 trillion cells in the adult human body has the same DNA text as was present in the original 23 pairs of chromosomes from the fertilized egg, one member of each pair having come from the mother and one from the father.
A second mechanism operating during the chromosome reduction process in sperm and egg cells further shuffles the genetic information inherited from mother
and father. In the first stage of the reduction process, each chromosome of a chromosome pair aligns with its partner. The maternally inherited chromosome 1 aligns with the paternally inherited chromosome 1, and so on through the 22 pairs; X chromosomes align with each other as well, but X and Y chromosomes do not. While the chromosome pairs are aligned, they exchange pieces to create new combinations. The recombined chromosomes are passed on in the sperm and eggs. As a consequence, the chromosomes we inherit from our parents are not exact copies of their chromosomes, but rather are mosaics of these parental chromosomes.
The swapping of material between chromosome pairs (as they align in the emerging sex cells) and the random selection (of half of each parent’s 46 chromosomes) in making sex cells is called recombination. Recombination is the principal source of diversity in individual human genomes.
The diverse variations occur both within the genes and in the regions of DNA sequences between the genes. A gene can be defined as a segment of DNA, usually from 1000 to 10,000 base pairs long, that “codes” for a protein. The cell produces specific proteins that correspond to the order of the base pairs (the “letters”) in the coding part of the gene.14 Human genes also contain noncoding sequences that regulate the cell type in which a protein will be synthesized and how much protein will be produced.15 Many genes contain interspersed noncoding, nonregulatory sequences that no longer participate in protein synthesis. These sequences, which have no apparent function, constitute about 23% of the base pairs within human genes.16 In terms of the metaphor of DNA as text, the gene is like an important paragraph in the book, often with some gibberish in it.
Proteins perform all sorts of functions in the body and thus produce observable characteristics. For example, a tiny part of the sequence that directs the production of the human group-specific complement protein (a protein that binds to vitamin D and transports it to certain tissues) is
G C A A A A T T G C C T G A T G C C A C A C C C A A G G A A C T G G C A.
14. The sequence in which the building blocks (amino acids) of a protein are arranged corresponds to the sequence of base pairs within a gene. (A sequence of three base pairs specifies a particular 1 of the 20 possible amino acids in the protein. The mapping of a set of three nucleotide bases to a particular amino acid is the genetic code. The cell makes the protein through intermediate steps involving coding RNA transcripts.) About 1.5% of the human genome codes for the amino acid sequences.
15. These noncoding but functional sequences include promoters, enhancers, and repressors.
16. This gene-related DNA consists of introns (which interrupt the coding sequences, called exons, in genes and which are edited out of the RNA transcript for the protein), pseudogenes (evolutionary remnants of once-functional genes), and gene fragments. The idea of a gene as a block of DNA (some of which is coding, some of which is regulatory, and some of which is functionless) is an oversimplification, but it is useful enough here. See, e.g., Mark B. Gerstein et al., What Is a Gene, Post-ENCODE? History and Updated Definition, 17 Genome Res. 669 (2007).
This gene always is located at the same position, or locus, on chromosome 4. As we have seen, most individuals have two copies of each gene at a given locus—one from the father and one from the mother.
A locus where almost all humans have the same DNA sequence is called monomorphic (“of one form”). A locus where the DNA sequence varies among significant numbers of individuals (more than 1% or so of the population possesses the variant) is called polymorphic (“of many forms”), and the alternative forms are called alleles. For example, the GC protein gene sequence has three common alleles that result from substitutions in a base at a given point. Where an A appears in one allele, there is a C in another. The third allele has the A, but at another point a G is swapped for a T. These changes are called single nucleotide polymorphisms (SNPs, pronounced “snips”).
If a gene is like a paragraph in a book, a SNP is a change in a letter somewhere within that paragraph (a substitution, a deletion, or an insertion), and the two versions of the gene that result from this slight change are the alleles. An individual who inherits the same allele from both parents is called a homozygote. An individual with distinct alleles is a heterozygote.
DNA sequences used for forensic analysis usually are not genes. They lie in the vast regions between genes (about 75% of the genome is extragenic) or in the apparently nonfunctional regions within genes. These extra- and intragenic regions of DNA have been found to contain considerable sequence variation, which makes them particularly useful in distinguishing individuals. Although the terms “locus,” “allele,” “homozygous,” and “heterozygous” were developed to describe genes, the nomenclature has been carried over to describe all DNA variation—coding and noncoding alike. Both types are inherited from mother and father in the same fashion.
B. What Are DNA Polymorphisms and How Are They Detected?
By determining which alleles are present at strategically chosen loci, the forensic scientist ascertains the genetic profile, or genotype, of an individual (at those loci). Although the differences among the alleles arise from alterations in the order of the ATGC letters, genotyping does not necessarily require “reading” the full DNA sequence. Here we outline the major types of polymorphisms that are (or could be) used in identity testing and the methods for detecting them.
Researchers are investigating radically new and efficient technologies to sequence entire genomes, one base pair at a time, but the direct sequencing methods now in existence are technically demanding, expensive, and time-consuming for whole-genome sequencing. Therefore, most genetic typing focuses on identifying only
those variations that define the alleles and does not attempt to “read out” each and every base pair as it appears. The exception is mitochondrial DNA, described in Section V. As next-generation sequencing technologies are perfected, however (see infra Section II.F), this situation could change.
2. Sequence-specific probes and SNP chips
Simple sequence variation, such as that for the GC locus, is conveniently detected using sequence-specific oligonucleotide (SSO) probes. A probe is a short, single strand of DNA. With GC typing, for example, probes for the three common alleles are attached to designated locations on a membrane. Copies of the variable sequence region of the GC gene in the crime scene sample are made with the polymerase chain reaction (PCR), which is discussed in the next section. These copies (in the form of single strands) are poured onto the membrane. Whichever allele is present in a single-stranded DNA fragment will cause the fragment to stick to the corresponding, immobilized probe strands. To permit the fragments of this type to be seen, a chemical “label” that catalyzes a color change at the spot where the DNA binds to its probe can be attached when the copies are made. A colored spot showing that the allele is present thus should appear on the membrane at the location of the probe that corresponds to this particular allele. If only one allele is present in the crime scene DNA (because of homozygosity), there will be no change at the spots where the other probes are located. If two alleles are present (heterozygosity), the corresponding two spots will change color.
This approach can be miniaturized and automated by embedding probes for many loci on a silicon chip. Commercially available “SNP chips” for disease research incorporate enough different probes to detect on the order of a million different known SNPs throughout the human genome. These chips have become a basic tool in searches for genetic changes associated with human diseases. They are described further in Section II.F.
Another category of DNA variations comes from the insertion of a variable number of tandem repeats (VNTR) at a locus. These were the first polymorphisms to find widespread use in identity testing and hence were the subject of most of the court opinions on the admissibility of DNA in the late 1980s and early 1990s. The core unit of a VNTR is a particular short DNA sequence that is repeated many times end-to-end. The first VNTRs to be used in genetic and forensic testing had core repeat sequences of 15–35 base pairs. In this testing, bacterial enzymes (known as “restriction enzymes”) were used to cut the DNA molecule both before and after the VNTR sequence. A small number of repeats in the VNTR region gives rise to a small “restriction fragment,” and a large number of repeats yields a large fragment. A substantial quantity of DNA from a crime scene sample is required to give a detectable number of VNTR fragments with this procedure.
The detection is accomplished by applying a probe that binds when it encounters the repeated core sequence. A radioactive or fluorescent molecule attached to the probe provides a way to mark the VNTR fragment. The probe ignores DNA fragments that do not include the VNTR core sequence. (There are many of these unwanted fragments, because the restriction enzymes chop up the DNA throughout the genome—not just at the VNTR loci.) The restriction fragments are sorted by a process known as electrophoresis, which separates DNA fragments based on size. Many early court opinions refer to this process as RFLP testing.17
Although RFLP-VNTR profiling is highly discriminating,18 it has several drawbacks. Not only does it require a substantial sample size, but it also is time-consuming and does not measure the fragment lengths to the nearest number of repeats. The measurement error inherent in the form of electrophoresis used (known as “gel electrophoresis”) is not a fundamental obstacle, but it complicates the determination of which profiles match and how often other profiles in the population would be declared to match.19 Consequently, forensic scientists have moved from VNTRs to another form of repetitive DNA known as short tandem repeats (STRs) or microsatellites. STRs have very short core repeats, two to seven base pairs in length, and they typically extend for only some 50 to 350 base pairs.20 Like the larger VNTRs, which extend for thousands of base pairs, STR sequences do not code for proteins, and the ones used in identity testing convey little or no information about an individual’s propensity for disease.21 Because STR alleles
17. It would be clearer to call it RFLP-VNTR testing, because the fragments being measured contain the VNTRs rather than some simpler polymorphisms that were used in genetic research and disease testing. A more detailed exposition of the steps in RFLP-VNTR profiling (including gel electrophoresis, Southern blotting, and autoradiography) can be found in the previous edition of this guide and in many judicial opinions circa 1990.
18. Alleles at VNTR loci generally are too long to be measured precisely by electrophoretic methods—alleles differing in size by only a few repeat units may not be distinguished. Although this makes for complications in deciding whether two length measurements that are close together result from the same allele, these loci are quite powerful for the genetic differentiation of individuals, because they tend to have many alleles that occur relatively rarely in the population. At a locus with only 20 such alleles (and most loci typically have many more), there are 210 possible genotypes. With five such loci, the number of possible genotypes is 2105, which is more than 400 billion.
19. For a case reversing a conviction as a result of an expert’s confusion on this score, see People v. Venegas, 954 P.2d 525 (Cal. 1998). More suitable procedures for match windows and probabilities are described in NRC II, supra note 9.
20. The numbers, and the distinction between “minisatellites” (VNTRs) and microsatellites (STRs), are not precise, but the mechanisms that give rise to the shorter tandem repeats differ from those that produce the longer ones. See Benjamin Lewin, Genes IX 124–25 (9th ed. 2008).
21. See David H. Kaye, Please, Let’s Bury the Junk: The CODIS Loci and the Revelation of Private Information, 102 Nw. U. L. Rev. Colloquy 70 (2007), available at http://www.law.northwestern.edu/lawreview/colloquy/2007/25/.
are much smaller than VNTR alleles, however, they can be amplified with PCR designed to copy only the locus of interest. This obviates the need for restriction enzymes, and it allows laboratories to analyze STR loci much more quickly. Because the amplified fragments are shorter, electrophoretic detection permits the exact number of base pairs in an STR to be determined, allowing alleles to be defined as discrete entities. Figure 2 illustrates the nature of allelic variation at an STR locus found on chromosome 16.
Figure 2. Three alleles of the D16S539 STR. The core sequence is GATA. The first allele listed has 9 tandem repeats, the second has 10, and the third has 11. The locus has other alleles (different numbers of repeats), shown in Figure 4.
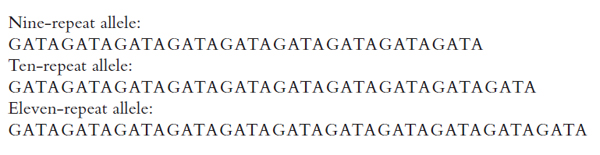
Although there are fewer alleles per locus for STRs than for VNTRs, there are many STRs, and they can be analyzed simultaneously. Such “multiplex” systems now permit the simultaneous analysis of 16 loci. A subset of 13 is standard in the United States (see infra Section II.D), and these are capable of distinguishing among almost everyone in the population.22
DNA contains the genetic information of an organism. In humans, most of the DNA is found in the cell nucleus, where it is organized into separate chromosomes. Each chromosome is like a book, and each cell has the same library (genome) of books of various sizes and shapes. There are two copies of each book of a particular size and shape, one that came from the father, the other from the mother. Thus, there are two copies of the book entitled “Chromosome One,” two copies of “Chromosome Two,” and so on. Genes are the most meaningful paragraphs in the books. Other parts of the text appear to have no coherent message. Two individuals sometimes have different versions (alleles) of the same paragraph. Some alleles result from the substitution of one letter for another. These are SNPs. Others come about from the insertion or deletion of single letters, and still
22. Usually, there are between 7 and 15 STR alleles per locus. Thirteen loci that have 10 STR alleles each can give rise to 5513, or 42 billion trillion, possible genotypes.
others represent a kind of stuttering repetition of a string of extra letters. These are the VNTRs and STRs.23 The locations within a chromosome where these interpersonal variations occur are called loci.
C. How Is DNA Extracted and Amplified?
DNA usually can be found in biological materials such as blood, bone, saliva, hair, semen, and urine. A combination of routine chemical and physical methods permits DNA to be extracted from cell nuclei and isolated from the other chemicals in a sample. PCR then is used to make exponentially large numbers of copies of targeted regions of the extracted DNA. PCR might be applied to the double-stranded DNA segments extracted and purified from a forensic sample as follows: First, the purified DNA is separated into two strands by heating it to near the boiling point of water. This “denaturing” takes about a minute. Second, the single strands are cooled, and “primers” attach themselves to the points at which the copying will start and stop. (Primers are small, manmade pieces of DNA, usually between 15 and 30 nucleotides long, of known sequences. If a locus of interest starts near the sequence ATCGAATCGGTAGCCATATG on one strand, a suitable primer would have the complementary sequence TAGCTTAGCCATCGGTATAC.) “Annealing” these primers takes about 45 seconds. Finally, the soup containing the annealed DNA strands, the enzyme DNA polymerase, and lots of the four nucleotide building blocks (A, C, G, and T) is warmed to a comfortable working temperature for the polymerase to insert the complementary base pairs one at a time, building a matching second strand bound to the original “template” and thus replicating part of the DNA strand that was separated from its partner in the first step. The same replication occurs with the separated partner as the template. This “extension” step for both templates takes about 2 minutes. The result is two identical double-stranded DNA segments, one made from each strand of the original DNA. The three-step cycle is repeated, usually 20 to 35 times in automated machines known as thermocyclers. Ideally, the first cycle results in two double-stranded DNA segments. The second cycle produces four, the third eight, and so on, until the number of copies of the original DNA is enormous. In practice, there is some inefficiency in the doubling process, but the yield from a 30-cycle amplification is generally about 1 million to 10 million copies of the targeted sequence.24 In this way, PCR magnifies short sequences of interest in a small number of DNA fragments into millions of exact copies. Machines that automate the PCR process are commercially available.
For PCR amplification to work properly and yield copies of only the desired sequence, however, care must be taken to achieve the appropriate chemical con-
23. In addition to the 23 pairs of books in the cell nucleus, other scraps of text reside in each of the mitochondria, the power plants of the cell. See infra Section V.
24. NRC II, supra note 9, at 69–70.
ditions and to avoid excessive contamination of the sample. A laboratory should be able to demonstrate that it can amplify targeted sequences faithfully with the equipment and reagents that it uses and that it has taken suitable precautions to avoid or detect contamination from foreign DNA. With small samples, it is possible that some alleles will be amplified and others missed (preferential amplification, discussed infra Section III.A.1), and mutations in the region of a primer can prevent the amplification of the allele downstream of the primer (null alleles).25
D. How Is STR Profiling Done with Capillary Electrophoresis?
In the most commonly used analytical method for detecting STRs, the STR fragments in the sample are amplified using primers with fluorescent tags. Each new STR fragment made in a PCR cycle bears a fluorescent dye. When struck by a source light, each dye glows with a particular color. The fragments are separated according to their length by electrophoresis in automated “genetic analyzer” machinery—a byproduct of the technology developed for the Human Genome Project that first sequenced most of the entire genome. In these machines, a long, narrow tube (a “capillary”) is filled with an entangled polymer or comparable sieving medium, and an electric field is applied to pull DNA fragments placed at one end of the tube through the medium. Shorter fragments slip through the medium more quickly than larger, bulkier ones. A laser beam is sent through a small glass window in the tube. The laser light excites the dye, causing it to fluoresce at a characteristic wavelength as the tagged fragments pass under the light. The intensity of the light emitted by the dye is recorded by a kind of electronic camera and transformed into a graph (an electropherogram), which shows a peak as an STR flashes by. A shorter allele will pass by the window and fluoresce first; a longer fragment will come by later, giving rise to another peak on the graph. Figure 3 provides a sketch of how the alleles with five and eight repeats of the GATA sequence at the D16S539 STR locus might appear in an electropherogram.
Medical and human geneticists were interested in STRs as markers in family studies to locate the genes that are associated with inherited diseases, and papers on their potential for identity testing appeared in the early 1990s. Developmental research to pick suitable loci moved into high gear in England, Europe, and Canada. Britain’s Forensic Science Service applied a four-locus testing system in 1994. Then it introduced the “second generation multiplex” (SGM)—for simultaneously typing six loci in 1996. These soon would be used to build England’s National DNA Database. The database system allows a computer to check the STR types of millions of known or suspected criminals against thousands of crime
25. A null allele will not lead to a false exclusion if the two DNA samples from the same individual are amplified with the same primer system, but it could lead to an exclusion at one locus when searching a database of STR profiles if the database profile was determined with a different PCR kit than the one used to analyze the crime scene DNA.
Figure 3. Sketch of an electropherogram for two D16S539 alleles. One allele has five repeats of the sequence GATA; the other has eight. Each GATA repeat is depicted as a small rectangle. Although only one copy of each allele (with a fluorescent molecule, or “tag” attached) is shown here, PCR generates a great many copies from the DNA sample with these alleles at the D16S539 locus. These copies are drawn through the capillary tube, and the tags glow as the STR fragments move through the laser beam. An electronic camera measures the colored light from the tags. Finally, a computer processes the signal from the camera to produce the electropherogram. Source: David H. Kaye, The Double Helix and the Law of Evidence 189, fig. 9.1 (2010).

scene samples. A six-locus STR profile can be represented as a string of 12 digits; each digit indicates the number of repeat units in the alleles at each locus. These discrete, numerical DNA profiles are far easier to compare mechanically than the complex patterns of fingerprints. In the United States, the FBI settled on 13 “core loci” to use in the U.S. national DNA database system. These are often called the “CODIS core loci,” and an additional 7 STR loci are under consideration.26
Modern genetic analyzers produce electropherograms for many loci at once. This “multiplexing” is accomplished by using dyes that fluoresce at distinct colors
26. Douglas R. Hares, Expanding the CODIS Core Loci in the United States, Forensic Sci. Int’l: Genetics (forthcoming 2011). CODIS stands for “convicted offender DNA index system.”
to label the alleles from different groups of loci. A separate set of fragments of known sizes that comigrate through the capillary function as a kind of ruler (an “internal-lane size standard”) to determine the lengths of the allelic fragments. Software processes the raw data to generate an electropherogram of the separate allele peaks of each color. By comparing the positions of the allele peaks to the size standard, the program determines the number of repeats in each allele. The plotted heights of the peaks (measured in relative fluorescent units, or RFUs) are proportional to the amount of the PCR product.
Figure 4 is an electropherogram of all 203 major alleles at 15 STR loci that can be typed in a single “multiplex” PCR reaction. (In addition, it shows the two alleles of the gene used to determine the sex of the contributor of a DNA
Figure 4. Alleles of 15 STR loci and the amelogenin sex-typing test from the AmpFISTR Identifiler kit. The bottom panel is a “sizing standard”—a set of peaks from DNA sequences of known lengths (in base pairs). The numbers in the vertical axis in each panel are relative fluorescence units (RFUs) that indicate the amount of light emitted after the laser beam strikes the fluorescent tag on an STR fragment.
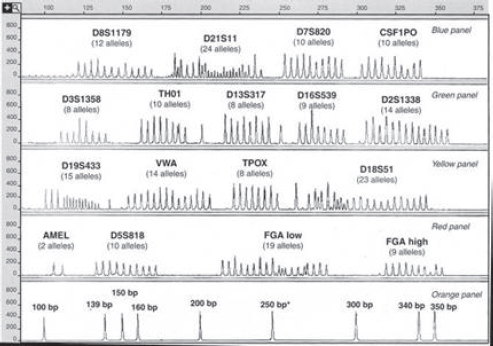
Note: Applied Biosystems makes the kit that produced these allelic ladders.
Source: John M. Butler, Forensic DNA Typing: Biology, Technology, and Genetics of STR Markers 128 (2d ed. 2005), Copyright Elsevier 2005, with the permission of Elsevier Academic Press. John Butler supplied the illustration.
sample.27) An electropherogram from an individual’s DNA would have only one or two peaks at each of these 15 STR loci (depending on whether the person is homozygous or heterozygous). These “allelic ladders” aid in deciding which allele a peak from an unknown sample represents.
Figure 5 is an electropherogram from the vaginal epithelial cells of the body of a girl who had been sexually assaulted and killed in People v. Pizarro.28 It was produced for the retrial in 2008 of the defendant who was linked to the victim by VNTR typing at his first trial in 1990.
Figure 5. Electropherogram for nine STR loci of the victim’s DNA in People v. Pizzaro. (The amelogenin locus and a sizing standard at the bottom also are included.) Some STR loci have small peaks, indicating that there was not much PCR product for those loci, likely because of DNA degradation. All of the STR loci have two peaks, as would be expected when the source is heterozygous at those loci.

Source: Steven Myers and Jeanette Wallin, California Department of Justice, provided the image.
27. The amelogenin gene, which is found on the X and the Y chromosomes, codes for a protein that is a major component of tooth enamel matrix. The copy on the X chromosome is 112 bp long. The copy on the Y chromosome has a string of six base pairs deleted, making it slightly shorter (106 bp). A female (XX) will have one peak at 112 bp. A male (XY) will have two peaks (at 106 and 112 bp).
28. 12 Cal. Rptr. 2d 436 (Ct. App. 1992), after remand, 3 Cal. Rptr. 3d 21 (Ct. App. 2003), review denied (Oct 15, 2003).
E. What Can Be Done to Validate a Genetic System for Identification?
Regardless of the kind of genetic system used for typing—STRs, SNPs, or still other polymorphisms—some general principles and questions can be applied to each system that is offered for courtroom use. First, the nature of the polymorphism should be well characterized. Is it a simple sequence polymorphism or a fragment length polymorphism? This information should be in the published literature or in archival genome databanks.
Second, the published scientific literature can be consulted to verify claims that a particular method of analysis can produce accurate profiles under various conditions. Although such validation studies have been conducted for all the systems ordinarily used in forensic work, determining the point at which the empirical validation of a particular system is sufficiently convincing to pass scientific muster may well require expert assistance.
Finally, the population genetics of the system should be characterized. As new systems are discovered, researchers typically analyze convenient collections of DNA samples from various human populations and publish studies of the relative frequencies of each allele in these population samples. These studies measure the extent of genetic variability at the polymorphic locus in the various populations, and thus of the potential probative power of the marker for distinguishing among individuals.
At this point, the capability of PCR-based procedures to ascertain DNA genotypes accurately cannot be doubted. Of course, the fact that scientists have shown that it is possible to extract DNA, to amplify it, and to analyze it in ways that bear on the issue of identity does not mean that a particular laboratory has adopted a suitable protocol and is proficient in following it. These case-specific issues are considered in Sections III and IV.
F. What New Technologies Might Emerge?
1. Miniaturized “lab-on-a-chip” devices
Miniaturized capillary electrophoresis (CE) devices have been developed for rapid detection of STRs (described in Section II.D) and other genetic analyses. The mini-CE systems consist of microchannels roughly the diameter of a hair etched on glass wafers (“chips”) using technology borrowed from the computer industry. The principles of electrophoretic separation are the same as with conventional CE systems. With microfluidic technologies, it is possible to integrate DNA extraction and PCR amplification processes with the CE separation in a single device, a so-called lab on a chip. Once a sample is added to the device, all the analytical steps are performed on the chip without further human contact. These integrated devices combine the benefits of simplified sample handling with rapid analysis
and are under active development for point-of-care medical diagnostics.29 Efforts are under way to develop an integrated microdevice for STR analysis that would improve the speed and efficiency of forensic DNA profiling. A portable device for rapid and secure analysis of samples in the field is a distinct possibility.30
The initial success of the Human Genome Project and the promise of “personalized medicine” is driving research to develop technologies for DNA analysis that are faster, cheaper, and less labor intensive. In 2004, the National Human Genome Research Institute announced funding for research leading to the “$1000 genome,” an achievement that would permit sequencing an individual’s genome for medical diagnosis and improved drug therapies. Advances in the years since 2004 suggest that this goal will be achieved before the target date of 2014,31 and the successful innovations could provide major advances in forensic DNA testing. However, it is too soon to identify which of the nascent sequencing technologies might emerge from the pack.
As of 2009, three different next-generation sequencing technologies were commercially available, and more instruments are in the pipeline.32 These new technologies generate massive amounts of DNA sequence data (100 million to 1 billion base pairs per run) at very low cost (under $50 per megabase). They do so by simultaneously sequencing millions of short fragments, then applying bioinformatics software to assemble the sequences in the correct order. These high-throughput sequencing technologies have demonstrated their usefulness in research applications. Two of these applications, the analysis of highly degraded DNA33 and the identification of microbial bioterrorism agents, are of forensic relevance.34 As the speed and cost of sequencing diminish and the necessary bioinformatics software becomes more accessible and effective, full-genome sequence
29. P. Yager et al., Microfluidic Diagnostic Technologies for Global Public Health, 442 Nature 412 (2006).
30. K.M. Horsman et al., Forensic DNA Analysis on Microfluidic Devices: A Review, 52 J. Forensic Sci. 784 (2007). As indicated in this review, there remain challenges to overcome before the forensic lab on a chip comes to fruition. However, given the progress being made on multiple research fronts in chip fabrication design and in microfluidic technology, these challenges seem surmountable.
31. R.F. Service, The Race for the $1000 Genome, 311 Science 1544 (2006).
32. Michael L. Metzker, Sequencing Technologies—The Next Generation, 11 Nature Rev. Genetics 31 (2010).
33. The next-generation technologies have been used to sequence highly degraded DNA from Neanderthal bones and from the hair of the extinct woolly mammoth. R.E. Green et al., Analysis of One Million Base Pairs of Neanderthal DNA, 444 Nature 330 (2006); W. Miller, Sequencing the Nuclear Genome of the Extinct Woolly Mammoth, 456 Nature 387 (2008). The approaches used in these studies are readily translatable to SNP typing of highly degraded DNA such as found in cases involving victims of mass disasters.
34. By sequencing entire bacterial genomes, researchers can rapidly differentiate organisms that have been genetically modified for biological warfare or terrorism from routine clinical and envi-
analysis or something approaching it could become a practical tool for human identification.
Hybridization microarrays are the third technological innovation with readily foreseeable forensic application. A microarray consists of a two-dimensional grid of many thousands of microscopic spots on a glass or plastic surface, each containing many copies of a short piece of single-stranded DNA tethered to the surface at one end; each spot can be thought of as a dense cluster of tiny, single-stranded DNA “whiskers” with their own particular sequence. A solution containing single-stranded target DNA is washed over the microarray surface. The whiskers on the array serve as probes to detect DNA (or RNA) with the corresponding complementary sequence. The spots that capture target DNA are identified, indicating the presence of that sequence in the target sample. (The hybridization can be detected in several different ways.) Microarrays are commercially available for the detection of SNPs in the human genome and for sequencing human mitochondrial DNA.35
4. What questions do the new technologies raise?
As these or other emerging technologies are introduced in court, certain basic questions will need to be answered. What is the principle of the new technology? Is it simply an extension of existing technologies, or does it invoke entirely new concepts? Is the new technology used in research or clinical applications independent of forensic science? Does the new technology have limitations that might affect its application in the forensic sphere? Finally, what testing has been done and with what outcomes to establish that the new technology is reliable when used on forensic samples? For next-generation sequencing technologies and microarray technologies, the questions may be directed as well to the bioinformatics methods used to analyze and interpret the raw data. Obtaining answers to these questions would likely require input both from experts involved in technology development and application and from knowledgeable forensic experts.
ronmental strains. B. La Scola et al., Rapid Comparative Genomic Analysis for Clinical Microbiology: The Francisella Tularensis Paradigm, 18 Genome Res. 742 (2008).
35. One study of 3000 Europeans used a commercial microarray with over half a million SNPs “to infer [the individuals’] geographic origin with surprising accuracy—often to within a few hundred kilometers.” John Novembre et al., Genes Mirror Geography Within Europe, 456 Nature 98, 98 (2008). Microarrays also are used in studies of variation in the number of copies of certain genes in different people’s genomes (copy number variation). Microarrays to detect pathogens and other targets also have been developed.
III. Sample Collection and Laboratory Performance
A. Sample Collection, Preservation, and Contamination
The primary determinants of whether DNA typing can be done on any particular sample are (1) the quantity of DNA present in the sample and (2) the extent to which it is degraded. Generally speaking, if a sufficient quantity of reasonable quality DNA can be extracted from a crime scene sample, no matter what the nature of the sample, DNA typing can be done without problem. Thus, DNA typing has been performed on old blood stains, semen stains, vaginal swabs, hair, bone, bite marks, cigarette butts, urine, and fecal material. This section discusses what constitutes sufficient quantity and reasonable quality in the context of STR typing. Complications from contaminants and inhibitors also are discussed. The special technique of mitotyping and the treatment of samples that contain DNA from two or more contributors are discussed in Section V.
1. Did the sample contain enough DNA?
Amounts of DNA present in some typical kinds of samples vary from a trillionth or so of a gram for a hair shaft to several millionths of a gram for a postcoital vaginal swab. Most PCR test protocols recommend samples on the order of 1 billionth to 5 billionths of a gram for optimum yields. Normally, the number of amplification cycles for nuclear DNA is limited to 28 or so to ensure that there is no detectable product for samples containing less than about 20 cell equivalents of DNA.36
Procedures for typing still smaller samples—down to a single cell’s worth of nuclear DNA—have been studied. These have been shown to work, to some extent, with trace or contact DNA left on the surface of an object such as the steering wheel of a car. The most obvious strategy is to increase the number of amplification cycles. The danger is that chance effects might result in one allele being amplified much more than another. Alleles then could drop out, small peaks from unusual alleles at other loci might “drop in,” and a bit of extraneous DNA could contribute to the profile. Other protocols have been developed for typing such “low copy number” (LCN) or “low template” (LT) DNA.37 LT-STR
36. This is about 100 to 200 trillionths of a gram. A lower limit of about 10 to 15 cells’ worth of DNA has been determined to give balanced amplification.
37. See, e.g., John Buckleton & Peter Gill, Low Copy Number, in Forensic DNA Evidence Interpretation 275 (John S. Buckleton et al. eds., 2005); Pamela J. Smith & Jack Ballantyne, Simplified Low-Copy-Number DNA Analysis by Post-PCR Purification, 52 J. Forensic Sci. 820 (2007).
profiles have been admitted in courts in a few countries,38 and they are beginning to appear in prosecutions in the United States.39
Although there are tests to estimate the quantity of DNA in a sample, whether a particular sample contains enough human DNA to allow typing cannot always be predicted accurately. The best strategy is to try. If a result is obtained, and if the controls (samples of known DNA and blank samples) have behaved properly, then the sample had enough DNA. The appearance of the same peaks in repeated runs helps assure that these alleles are present.40
2. Was the sample of sufficient quality?
The primary determinant of DNA quality for forensic analysis is the extent to which the long DNA molecules are intact. Within the cell nucleus, each molecule of DNA extends for millions of base pairs. Outside the cell, DNA spontaneously degrades into smaller fragments at a rate that depends on temperature, exposure to oxygen, and, most importantly, the presence of water.41 In dry biological samples, protected from air, and not exposed to temperature extremes, DNA degrades very slowly. STR testing has proved effective with old and badly degraded material such as the remains of the Tsar Nicholas family (buried in 1918 and recovered in 1991).42
38. E.g., R. v. Reed [2009] (CA Crim. Div.) EWCA Crim. 2698, ¶ 74 (reviewing expert submissions and concluding that “Low Template DNA can be used to obtain profiles capable of reliable interpretation if the quantity of DNA that can be analysed is above the stochastic threshold [of] between 100 and 200 picograms”).
39. People v. Megnath, 898 N.Y.S.2d 408 (N.Y. Sup. Ct. 2010) (reasoning that “LCN DNA analysis” uses the same steps as STR analysis of larger samples and that the modifications in the procedure used by the laboratory in the case were generally accepted); cf. United States v. Davis, 602 F. Supp. 2d 658 (D. Md. 2009) (avoiding “making a finding with regard to the dueling definitions of LCN testing advocated by the parties” by finding that “the amount of DNA present in the evidentiary samples tested in this case” was in the normal range). These cases and the admissibility of low-template DNA analysis are discussed in Kaye et al., supra note 1, § 9.2.3(c).
40. John M. Butler & Cathy R. Hill, Scientific Issues with Analysis of Low Amounts of DNA, LCN Panel on Scientific Issues with Low Amounts of DNA, Promega Int’l Symposium on Human Identification, Oct. 15, 2009, available at http://www.cstl.nist.gov/strbase/pub_pres/Butler_Promega2009-LCNpanelfor-STRBase.pdf.
41. Other forms of chemical alteration to DNA are well studied, both for their intrinsic interest and because chemical changes in DNA are a contributing factor in the development of cancers in living cells. Some forms of DNA modification, such as that produced by exposure to ultraviolet radiation, inhibit the amplification step in PCR-based tests, whereas other chemical modifications appear to have no effect. C.L. Holt et al., TWGDAM Validation of AmpFlSTR PCR Amplification Kits for Forensic DNA Casework, 47 J. Forensic Sci. 66 (2002); George F. Sensabaugh & Cecilia von Beroldingen, The Polymerase Chain Reaction: Application to the Analysis of Biological Evidence, in Forensic DNA Technology 63 (Mark A. Farley & James J. Harrington eds., 1991).
42. Peter Gill et al., Identification of the Remains of the Romanov Family by DNA Analysis, 6 Nature Genetics 130 (1994).
The extent to which degradation affects a PCR-based test depends on the size of the DNA segment to be amplified. For example, in a sample in which the bulk of the DNA has been degraded to fragments well under 1000 base pairs in length, it may be possible to amplify a 100-base-pair sequence, but not a 1000-base-pair target. Consequently, the shorter alleles may be detected in a highly degraded sample, but the larger ones may be missed. Fortunately, the size differences among STR alleles at a locus are quite small (typically no more than 50 base pairs). Therefore, if there is a degradation effect on STR typing, it is usually “locus dropout”—in cases involving severe degradation, loci yielding larger products (greater than 200 base pairs) may not be detected.43
DNA can be exposed to a great variety of environmental insults without any effect on its capacity to be typed correctly. Exposure studies have shown that contact with a variety of surfaces, both clean and dirty, and with gasoline, motor oil, acids, and alkalis either have no effect on DNA typing or, at worst, render the DNA untypable.44
Although contamination with microbes generally does little more than degrade the human DNA, other problems sometimes can occur. Therefore, the validation of DNA typing systems should include tests for interference with a variety of microbes to see if artifacts occur. If artifacts are observed, then control tests should be applied to distinguish between the artifactual and the true results.
1. What forms of quality control and assurance should be followed?
DNA profiling is valid and reliable, but confidence in a particular result depends on the quality control and quality assurance procedures in the laboratory. Quality control refers to measures to help ensure that a DNA-typing result (and its interpretation) meets a specified standard of quality. Quality assurance refers to monitoring, verifying, and documenting laboratory performance. A quality assurance program helps demonstrate that a laboratory is meeting its quality control objectives and thus justifies confidence in the quality of its product.45
43. Holt et al., supra note 41. Special primers and very short STRs give better results with extremely degraded samples. See Michael D. Coble & John M. Butler, Characterization of New MiniSTR Loci to Aid Analysis of Degraded DNA, 50 J. Forensic Sci. 43 (2005).
44. Holt et al., supra note 41. Most of the effects of environmental insult readily can be accounted for in terms of basic DNA chemistry. For example, some agents produce degradation or damaging chemical modifications. Other environmental contaminants inhibit restriction enzymes or PCR. (This effect sometimes can be reversed by cleaning the DNA extract to remove the inhibitor.) But environmental insult does not result in the selective loss of an allele at a locus or in the creation of a new allele at that locus.
45. For a review of the history of quality assurance in forensic DNA testing, see J.L. Peterson et al., The Feasibility of External Blind DNA Proficiency Testing. I. Background and Findings, 48 J. Forensic Sci. 21, 22 (2003).
Professional bodies within forensic science have described procedures for quality assurance. Guidelines for DNA analysis have been prepared by FBI-appointed groups (the current incarnation is known as SWGDAM);46 a number of states require forensic DNA laboratories to be accredited;47 and federal law requires accreditation or other safeguards of laboratories that receive certain federal funds48 or participate in the national DNA database system.49
a. Documentation
Quality assurance guidelines normally call for laboratories to document laboratory organization and management, personnel qualifications and training, facilities, evidence control procedures, validation of methods and procedures, analytical procedures, equipment calibration and maintenance, standards for case documentation and report writing, procedures for reviewing case files and testimony, proficiency testing, corrective actions, audits, safety programs, and review of subcontractors.
46. The FBI established the Technical Working Group on DNA Analysis Methods (TWGDAM) in 1988 to develop standards. The DNA Identification Act of 1994, 42 U.S.C. § 14131(a) & (c) (2006), created a DNA Advisory Board (DAB) to assist in promulgating quality assurance standards, but the legislation allowed the DAB to expire after 5 years (unless extended by the Director of the FBI). 42 U.S.C. § 14131(b) (2008). TWGDAM functioned under DAB, 42 U.S.C. § 14131(a) (2006), and was renamed the Scientific Working Group on DNA Analysis Methods (SWGDAM) in 1999. When the FBI allowed DAB to expire, SWGDAM replaced DAB. See Norah Rudin & Keith Inman, An Introduction to Forensic DNA Analysis 180 (2d ed. 2002); Paul C. Giannelli, Regulating Crime Laboratories: The Impact of DNA Evidence, 15 J.L. & Pol’y 59, 82–83 (2007).
47. New York was the first state to impose this requirement. N.Y. Exec. Law § 995-b (McKinney 2006) (requiring accreditation by the state Forensic Science Commission).
48. The Justice for All Act, enacted in 2004, required DNA labs to be accredited within 2 years “by a nonprofit professional association of persons actively involved in forensic science that is nationally recognized within the forensic science community” and to “undergo external audits, not less than once every 2 years, that demonstrate compliance with standards established by the Director of the Federal Bureau of Investigation.” 42 U.S.C. § 14132(b)(2) (2006). Established in 1981, the American Society of Crime Laboratory Directors–Laboratory Accreditation Board (ASCLD-LAB) accredits forensic laboratories. Giannelli, supra note 46, at 75. The 2004 Act also requires applicants for federal funds for forensic laboratories to certify that the laboratories use “generally accepted laboratory practices and procedures, established by accrediting organizations or appropriate certifying bodies,” 42 U.S.C. § 3797k(2) (2004), and that “a government entity exists and an appropriate process is in place to conduct independent external investigations into allegations of serious negligence or misconduct substantially affecting the integrity of the forensic results committed by employees or contractors of any forensic laboratory system, medical examiner’s office, coroner’s office, law enforcement storage facility, or medical facility in the State that will receive a portion of the grant amount.” Id. § 3797k(4). There have been problems in implementing the § 3797k(4) certification requirement. See Office of the Inspector General, U.S. Dep’t of Justice, Review of the Office of Justice Programs’ Paul Coverdell Forensic Science Improvement Grants Program, Evaluation and Inspections Report I-2008-001 (2008), available at http://www.usdoj.gov/oig/reports/OJP/e0801/index.htm.
49. See 42 U.S.C § 14132 (b)(2) (2006) (requiring as of late 2006, that records in the database come from laboratories that “have been accredited by a nonprofit professional association…and…undergo external audits, not less than once every 2 years [and] that demonstrate compliance with standards established by the Director of the Federal Bureau of Investigation….”).
Of course, maintaining documentation and records alone does not guarantee the correctness of results obtained in any particular case. Errors in analysis or interpretation might occur as a result of a deviation from an established procedure, analyst misjudgment, or an accident. Although case review procedures within a laboratory should be designed to detect errors before a report is issued, it is always possible that some incorrect result will slip through. Accordingly, determination that a laboratory maintains a strong quality assurance program does not eliminate the need for case-by-case review.
b. Validation
The validation of procedures is central to quality assurance. Developmental validation is undertaken to determine the applicability of a new test to crime scene samples; it defines conditions that give reliable results and identifies the limitations of the procedure. For example, a new genetic marker being considered for use in forensic analysis will be tested to determine if it can be typed reliably in both fresh samples and in samples typical of those found at crime scenes. The validation would include testing samples originating from different tissues—blood, semen, hair, bone, samples containing degraded DNA, samples contaminated with microbes, samples containing DNA mixtures, and so on. Developmental validation of a new set of loci also includes the generation of population databases and the testing of alleles for statistical independence. Developmental validation normally results in publication in the scientific literature, but a new procedure can be validated in multiple laboratories well ahead of publication.
Internal validation, on the other hand, involves the capacity of a specific laboratory to analyze the new loci. The laboratory should verify that it can reliably perform an established procedure that already has undergone developmental validation. In particular, before adopting a new procedure, the laboratory should verify its ability to use the system in a proficiency trial.50
c. Proficiency testing
Proficiency testing in forensic genetic testing is designed to ascertain whether an analyst can correctly determine genetic types in a sample whose origin is unknown to the analyst but is known to a tester. Proficiency is demonstrated by making correct genetic typing determinations in repeated trials. The laboratory also can be tested to verify that it correctly computes random-match probabilities or similar statistics.
An internal proficiency trial is conducted within a laboratory. One person in the laboratory prepares the sample and administers the test to another person in the laboratory. In an external trial, the test sample originates from outside the labo-
50. Both forms of validation build on the accumulated body of knowledge and experience. Thus, some aspects of validation testing need be repeated only to the extent required to verify that previously established principles apply.
ratory—from another laboratory, a commercial vendor, or a regulatory agency. In a declared (or open) proficiency trial, the analyst knows the sample is a proficiency sample. The DNA Identification Act of 1994 requires proficiency testing for analysts in the FBI as well as those in laboratories participating in the national database or receiving federal funding,51 and the standards of accrediting bodies typically call for periodic open, external proficiency testing.52
In a blind (or, more properly, “full blind”) trial, the sample is submitted so that the analyst does not recognize it as a proficiency sample. A full-blind trial provides a better indication of proficiency because it ensures that the analyst will not give the trial sample any special attention, and it tests more steps in the laboratory’s processing of samples. However, full-blind proficiency trials entail considerably more organizational effort and expense than open proficiency trials. Obviously, the “evidence” samples prepared for the trial have to be sufficiently realistic that the laboratory does not suspect the legitimacy of the submission. A police agency and prosecutor’s office have to submit the “evidence” and respond to laboratory inquiries with information about the “case.” Finally, the genetic profile from a proficiency test must not be entered into regional and national databases. Consequently, although some forensic DNA laboratories participate in full-blind testing, they are not required to do so.53
2. How should samples be handled?
Sample mishandling, mislabeling, or contamination, whether in the field or in the laboratory, is more likely to compromise a DNA analysis than is an error in genetic typing. For example, a sample mixup due to mislabeling reference blood samples taken at the hospital could lead to incorrect association of crime scene samples to a reference individual or to incorrect exclusions. Similarly, packaging two items with wet bloodstains into the same bag could result in a transfer of stains between the items, rendering it difficult or impossible to determine whose blood was originally on each item. Contamination in the laboratory may result in artifactual
51. 42 U.S.C. § 14132(b)(2) (requiring external proficiency testing of laboratories for participation in the national database); id. § 14133(a)(1)(A) (2006) (same for FBI examiners).
52. See Peterson et al., supra note 45, at 24 (describing the ASCL-LAB standards). Certification by the American Board of Criminalistics as a specialist in forensic biology DNA analysis requires one proficiency trial per year. Accredited laboratories must maintain records documenting compliance with required proficiency test standards.
53. The DNA Identification Act of 1994 required the director of the National Institute of Justice to report to Congress on the feasibility of establishing an external blind proficiency testing program for DNA laboratories. 42 U.S.C. § 14131(c) (2006). A National Forensic DNA Review Panel advised the Director that “blind proficiency testing is possible, but fraught with problems” of the kind listed above). Peterson et al., supra note 46, at 30. It “recommended that a blind proficiency testing program be deferred for now until it is more clear how well implementation of the first two recommendations [the promulgation of guidelines for accreditation, quality assurance, and external audits of casework] are serving the same purposes as blind proficiency testing.” Id.
typing results or in the incorrect attribution of a DNA profile to an individual or to an item of evidence. Procedures should be prescribed and implemented to guard against such error.
Mislabeling or mishandling can occur when biological material is collected in the field, when it is transferred to the laboratory, when it is in the analysis stream in the laboratory, when the analytical results are recorded, or when the recorded results are transcribed into a report. Mislabeling and mishandling can happen with any kind of physical evidence and are of great concern in all fields of forensic science. Checkpoints should be established to detect mislabeling and mishandling along the line of evidence flow. Investigative agencies should have guidelines for evidence collection and labeling so that a chain of custody is maintained. Similarly, there should be guidelines, produced with input from the laboratory, for handling biological evidence in the field.
Professional guidelines and recommendations require documented procedures to ensure sample integrity and to avoid sample mixups, labeling errors, recording errors, and the like.54 They also mandate case review to identify inadvertent errors before a final report is released. Finally, laboratories must retain, when feasible, portions of the crime scene samples and extracts to allow reanalysis.55 However, retention is not always possible. For example, retention of original items is not to be expected when the items are large or immobile (e.g., a wall or sidewalk). In such situations, a swabbing or scraping of the stain from the item would typically be collected and retained. There also are situations where the sample is so small that it will be consumed in the analysis.
Assuming that appropriate chain-of-custody and evidence-handling protocols are in place, the critical question is whether there are deviations in the particular case. This may require a review of the total case documentation as well as the laboratory findings. In addition, the opportunity to retest original evidence items or the material extracted from them is an important safeguard against error because of mislabeling and mishandling. Should mislabeling or mishandling have occurred, reanalysis of the original sample and the intermediate extracts should detect not only the fact of the error but also the point at which it occurred.56
54. SWGDAM guidelines are published as FBI, Standards for Forensic DNA Testing Labs, available at http://www.fbi.gov/hq/lab/codis/forensic.htm (last visited Feb. 16, 2010).
55. Forensic laboratories have a professional responsibility to preserve retained evidence so as to minimize degradation. See id., standard 7.2.1. Furthermore, failure to preserve potentially exculpatory evidence has been treated as a denial of due process and grounds for suppression. People v. Nation, 604 P.2d 1051, 1054–55 (Cal. 1980). In Arizona v. Youngblood, 488 U.S. 51 (1988), however, the Supreme Court held that a police agency’s failure to preserve evidence not known to be exculpatory does not constitute a denial of due process unless “bad faith” can be shown. Ironically, DNA testing that was not available at Youngblood’s trial established that he had been falsely convicted. Maurice Possley, DNA Exonerates Inmate Who Lost Key Test Case: Prosecutors Ruined Evidence in Original Trial, Chi. Trib., Aug. 10, 2000, at 6.
56. Of course, retesting cannot correct all errors that result from mishandling of samples, but it is even possible in some cases to detect mislabeling at the point of sample collection if the genetic
Contamination describes any situation in which foreign material is mixed with a sample of DNA. As noted in Section III.A.2, contamination by non-biological materials, such as gasoline or grit, can cause test failures, but they are not a source of genetic typing errors. Similarly, contamination with nonhuman biological materials, such as bacteria, fungi, or plant materials, is generally not a problem. These contaminants may accelerate DNA degradation, but they do not generate spurious human genetic types.
The contamination of greatest concern is that resulting from the addition of human DNA. This sort of contamination can occur three ways. First, the crime scene samples by their nature may contain a mixture of fluids or tissues from different individuals. Examples include vaginal swabs collected as sexual assault evidence and bloodstain evidence from scenes where several individuals shed blood. Mixtures are the subject of Section V.C.
Second, the crime scene samples may be inadvertently contaminated in the course of sample handling in the field or in the laboratory. Inadvertent contamination of crime scene DNA with DNA from a reference sample could lead to a false inclusion.
Third, carryover contamination in PCR-based typing can occur if the amplification products of one typing reaction are carried over into the reaction mix for a subsequent PCR reaction. If the carryover products are present in sufficient quantity, they could be preferentially amplified over the target DNA. The primary strategy used in most forensic laboratories to protect against carryover contamination is to keep PCR products away from sample materials and test reagents by having separate work areas for pre-PCR and post-PCR sample handling, by preparing samples in controlled-air-flow biological safety hoods, by using dedicated equipment (such as pipetters) for each of the various stages of sample analysis, by decontaminating work areas after use (usually by wiping down or by irradiating with ultraviolet light), and by having a one-way flow of sample from the pre-PCR to post-PCR work areas. Additional protocols are used to detect any carryover contamination.57
In the end, whether a laboratory has conducted proper tests and whether it conducted them properly depends both on the general standard of practice and
typing results on a particular sample are inconsistent with an otherwise consistent reconstruction of events. For example, a mislabeling of husband and wife samples in a paternity case might result in an apparent maternal exclusion, a very unlikely event. The possibility of mislabeling could be confirmed by testing the samples for gender and ultimately verified by taking new samples from each party under better controlled conditions.
57. Standard protocols include the amplification of blank control samples—those to which no DNA has been added. If carryover contaminants have found their way into the reagents or sample tubes, these will be detected as amplification products. Outbreaks of carryover contamination can also be recognized by monitoring test results. Detection of an unexpected and persistent genetic profile in different samples indicates a contamination problem. When contamination outbreaks are detected, appropriate corrective actions should be taken, and both the outbreak and the corrective action should be documented.
on the questions posed in the particular case. There is no universal checklist, but the selection of tests and the adherence to the correct test procedures can be reviewed by experts and by reference to professional standards such as the SWGDAM guidelines.
IV. Inference, Statistics, and Population Genetics in Human Nuclear DNA Testing
The results of DNA testing can be presented in various ways. With discrete allele systems, such as STRs, it is natural to speak of “matching” and “nonmatching” profiles. If the genetic profile obtained from the biological sample taken from the crime scene or the victim (the “trace evidence sample”) matches that of a particular individual, that individual is included as a possible source of the sample. But other individuals also might possess a matching DNA profile. Accordingly, the expert should be asked to provide some indication of how significant the match is. If, on the other hand, the genetic profiles are different, then the individual is excluded as the source of the trace evidence. Typically, proof tending to show that the defendant is the source incriminates the defendant, whereas proof that someone else is the source exculpates the defendant.58 This section elaborates on these ideas, indicating issues that can arise in connection with an expert’s testimony interpreting the results of a DNA test.
A. What Constitutes a Match or an Exclusion?
When the DNA from the trace evidence clearly does not match the DNA sample from the suspect, the DNA analysis demonstrates that the suspect’s DNA is not in the forensic sample. Indeed, if the samples have been collected, handled, and analyzed properly, then the suspect is excluded as a possible source of the DNA in the forensic sample. As a practical matter, such exclusionary results normally would keep charges from being filed against the excluded suspect.
At the other extreme, the genotypes at a large number of loci can be clearly identical. In these cases, the DNA evidence is quite incriminating, and the challenge for the legal system lies in explaining just how probative it is. Naturally, as with exclusions, inclusions are most powerful when the samples have been
58. Whether being the source of the forensic sample is incriminating and whether someone else being the source is exculpatory depends on the circumstances. For example, a suspect who might have committed the offense without leaving the trace evidence sample still could be guilty. In a rape case with several rapists, a semen stain could fail to incriminate one assailant because insufficient semen from that individual is present in the sample.
collected, handled, and analyzed properly. But there is one logical difference between exclusions and inclusions. If it is accepted that the samples have different genotypes, then the conclusion that the DNA in them came from different individuals is essentially inescapable.59 In contrast, even if two samples have the same genotype, there is a chance that the forensic sample came not from the defendant, but from another individual who has the same genotype. This complication has produced extensive arguments over the statistical procedures for assessing this chance or related quantities. This problem of describing the significance of an unequivocal match is the subject of the remaining parts of this section.
Some cases lie between the poles of a clear inclusion or a definite exclusion. For example, when the trace evidence sample is small and extremely degraded, STR profiling can be afflicted with allelic “drop-in” and “drop-out,” requiring judgments as to whether true peaks are missing and whether spurious peaks are present. Experts then might disagree about whether a suspect is included or excluded—or whether any conclusion can be drawn.60
B. What Hypotheses Can Be Formulated About the Source?
If the defendant is the source of DNA of sufficient quantity and quality found at a crime scene, then a DNA sample from the defendant and the crime scene sample should have the same profile. The inference required in assessing the evidence, however, runs in the opposite direction. The forensic scientist reports that the sample of DNA from the crime scene and a sample from the defendant have the same genotype. The prosecution’s hypothesis is that the defendant is the source of the crime scene sample.61
Conceivably, other hypotheses could account for the matching profiles. One possibility is laboratory error—the genotypes are not actually the same even though the laboratory thinks that they are. This situation could arise from mistakes
59. The legal implications of this fact are discussed in Kaye et al., supra note 1, § 13.3.2.
60. See, e.g., State v. Murray, 174 P.3d 407, 417–18 (Kan. 2008) (inconclusive Y-STR results were presented as consistent with the defendant’s blood). Since the early days of DNA testing, concerns have been expressed about subjective aspects of specific procedures that leave room for “observer effects” in interpreting data. See William C. Thompson & Simon Ford, The Meaning of a Match: Sources of Ambiguity in the Interpretation of DNA Prints, in Forensic DNA Technology (M. Farley & J. Harrington eds., 1990); see generally D. Michael Risinger et al., The Daubert/Kumho Implications of Observer Effects in Forensic Science: Hidden Problems of Expectation and Suggestion, 90 Calif. L. Rev. 1 (2002). A number of commentators have proposed that the analyst determine the profile of a trace evidence sample before knowing the profile of any suspects. Dan E. Krane et al., Sequential Unmasking: A Means of Minimizing Observer Effects in Forensic DNA Interpretation, 53 J. Forensic Sci. 1006 (2008).
61. That the defendant is the source does not necessarily mean that the defendant is guilty of the offense charged. Aside from issues of intent or knowledge that have nothing to do with DNA, there remains, for example, the possibility that the two samples match because someone framed the defendant by putting a sample of defendant’s DNA at the crime scene or in the container of DNA thought to have come from the crime scene.
in labeling or handling samples or from cross-contamination of the samples. As the 1992 NRC report cautioned, “[e]rrors happen, even in the best laboratories, and even when the analyst is certain that every precaution against error was taken.”62
Another possibility is that the laboratory analysis is correct—the genotypes are truly identical—but the forensic sample came from another individual. In general, the true source might be a close relative of the defendant63 or an unrelated person who, as luck would have it, just happens to have the same profile as the defendant. The former hypothesis we shall refer to as kinship, and the latter as coincidence. To infer that the defendant is the source of the crime scene DNA, one must reject these alternative hypotheses of laboratory error, kinship, and coincidence. Table 1 summarizes the logical possibilities.
Table 1. Hypotheses That Might Explain a Match Between Defendant’s DNA and DNA at a Crime Scenea
| IDENTITY: | Same genotype, defendant’s DNA at crime scene |
| NONIDENTITY: | |
| Lab error | Different genotypes mistakenly found to be the same |
| Kinship | Same genotype, relative’s DNA at crime scene |
| Coincidence | Same genotype, unrelated individual’s DNA |
| aCf. N.E. Morton, The Forensic DNA Endgame, 37 Jurimetrics J. 477, 480 tbl. 1 (1997). | |
Some scientists have urged that probabilities associated with false-positive error, kinship, or coincidence be presented to juries. Although it is not clear that this goal is feasible, scientific knowledge and more conventional evidence can help in assessing the plausibility of these alternative hypotheses. If laboratory error, kinship, and coincidence are rejected as implausible, then only the hypothesis of identity remains. We turn, then, to the considerations that affect the chances of a match when the defendant is not the source of the trace evidence.
C. Can the Match Be Attributed to Laboratory Error?
Although many experts would concede that even with rigorous protocols, the chance of a laboratory error exceeds that of a coincidental match, quantifying the former probability is a formidable task. Some commentary proposes using the proportion of false positives that the particular laboratory has experienced in blind
62. NRC I, supra note 8, at 89.
63. A close relative, for these purposes, would be a brother, uncle, nephew, etc. For relationships more distant than second cousins, the probability of a chance match is nearly as small as for persons of the same ethnic subgroup. Bernard Devlin & Kathryn Roeder, DNA Profiling: Statistics and Population Genetics, in 1 Modern Scientific Evidence: The Law and Science of Expert Testimony § 18-3.1.3, at 724 (David L. Faigman et al. eds., 1997).
proficiency tests or the rate of false positives on proficiency tests averaged across all laboratories.64 Indeed, the 1992 NRC Report remarks that “proficiency tests provide a measure of the false-positive and false-negative rates of a laboratory.”65 Yet the same report recognizes that “errors on proficiency tests do not necessarily reflect permanent probabilities of false-positive or false-negative results,”66 and the 1996 NRC report suggests that a probability of a false-positive error that would apply to a specific case cannot be estimated objectively.67 If the false-positive probability were, say, 0.001, it would take tens of thousands of proficiency tests to estimate that probability accurately, and the application of an historical industry-wide error rate to a particular laboratory at a later time would be debatable.68
Most commentators who urge the use of proficiency tests to estimate the probability that a laboratory has erred in a particular case agree that blind proficiency testing cannot be done in sufficient numbers to yield an accurate estimate of a small error rate. However, they maintain that proficiency tests, blind or otherwise, should be used to provide a conservative estimate of the false-positive error probability.69 For example, if there were no errors in 100 tests, a 95% confidence interval would include the possibility that the error rate could be almost as high as 3%.70
Whether or not a case-specific probability of laboratory error can be estimated with proficiency tests, traditional legal and scientific procedures can help to assess the possibilities of errors in handling or analyzing the samples. Scrutinizing the chain of custody, examining the laboratory’s protocol, verifying that it adhered to that protocol, and conducting confirmatory tests (including testing by the defense) can help show that the profiles really do match.
D. Could a Close Relative Be the Source?
With enough loci to test, all individuals except identical twins should be distinguishable. With existing technology and small sample sizes of DNA recovered from crime scenes, however, this ideal is not always attainable. A thorough inves-
64. E.g., Jonathan J. Koehler, Error and Exaggeration in the Presentation of DNA Evidence at Trial, 34 Jurimetrics J. 21, 37–38 (1993).
65. NRC I, supra note 8, at 94.
66. Id. at 89.
67. NRC II, supra note 9, at 85–87.
68. Id. at 85–86; Devlin & Roeder, supra note 63, § 18-5.3, at 744–45. Such arguments have not persuaded the proponents of estimating the probability of error from industry-wide proficiency testing. E.g., Jonathan J. Koehler, Why DNA Likelihood Ratios Should Account for Error (Even When a National Research Council Report Says They Should Not), 37 Jurimetrics J. 425 (1997).
69. E.g., Jonathan J. Koehler, DNA Matches and Statistics: Important Questions, Surprising Answers, 76 Judicature 222, 228 (1993); Richard Lempert, After the DNA Wars: Skirmishing with NRC II, 37 Jurimetrics J. 439, 447–48, 453 (1997).
70. See NRC II, supra note 9, at 86 n.1. For an explanation of confidence intervals, see David H. Kaye & David A. Freedman, Reference Guide on Statistics, in this manual.
tigation might extend to all known relatives, but this is not feasible in every case, and there is always the chance that some unknown relatives are in the suspect population. Formulas are available for computing the probability that any person with a specified degree of kinship to the defendant also possesses the incriminating genotype. For example, the probability that an untested brother (or sister) would match at four loci (with alleles that each occur in 10% of the population) is about 1/380;71 the probability that an aunt (or uncle) would match is about 1/100,000.72
E. Could an Unrelated Person Be the Source?
Another rival hypothesis is coincidence: The defendant is not the source of the crime scene DNA but happens to have the same genotype as an unrelated individual who is the true source. Various procedures for assessing the plausibility of this hypothesis are available. In principle, one could test all conceivable suspects. If everyone except the defendant has a nonmatching profile, then the defendant must be the source. But exhaustive, error-free testing of the population of conceivable suspects is almost never feasible. The suspect population normally defies any enumeration, and in the typical crime where DNA evidence is found, the population of possible perpetrators is so huge that even if all of its members could be listed, they could not all be tested.73
An alternative procedure would be to take a sample of people from the suspect population, find the relative frequency of the profile in this sample, and use that statistic to estimate the frequency in the entire suspect population. The smaller the frequency, the less likely it is that the defendant’s DNA would match if the defendant were not the source of trace evidence. Again, however, the suspect population is difficult to define, so some surrogate must be used. The procedure commonly followed is to estimate the relative frequency of the incriminating
71. For a case with conflicting calculations of the probability of an untested brother having a matching genotype, see McDaniel v. Brown, 130 S. Ct. 665 (2010) (per curiam). The correct computation is given in David H. Kaye, “False, but Highly Persuasive”: How Wrong Were the Probability Estimates in McDaniel v. Brown? 108 Mich. L. Rev. First Impressions 1 (2009), available at http://www.michiganlawreview.org/assets/fi/108/kaye.pdf.
72. These figures follow from the equations in NRC II, supra note 9, at 113. The large discrepancy between two siblings on the one hand, and an uncle and nephew on the other, reflects the fact that the siblings have far more shared ancestry. All their genes are inherited through the same two parents. In contrast, a nephew and an uncle inherit from two unrelated mothers, and so will have few maternal alleles in common. As for paternal alleles, the nephew inherits not from his uncle, but from his uncle’s brother, who shares by descent only about one-half of his alleles with the uncle.
73. As the cost of DNA profiling drops, it will become technically and economically feasible to have a comprehensive, population-wide DNA database that could be used to produce a list of nearly everyone whose DNA profile is consistent with the trace evidence DNA. Whether such a system would be constitutionally and politically acceptable is another question. See David H. Kaye & Michael S. Smith, DNA Identification Databases: Legality, Legitimacy, and the Case for Population-Wide Coverage, 2003 Wis. L. Rev. 413.
genotype in a large population. But even this cannot be done directly because each possible multilocus profile is so rare that it is not likely to show up in any sample of a reasonable size. However, the frequencies of most alleles can be determined accurately by sampling the population to construct databases that reveal how often each allele occurs. Principles of population genetics then can be applied to combine the estimated allele frequencies into an estimate of the probability that a person born in the population will have the multilocus genotype. This probability often is referred to as the random-match probability. This section describes how the allele frequencies are estimated from samples and how the random-match probability is computed from allele frequencies.
1. Estimating allele frequencies from samples
As we saw in Section II.B, the loci currently used in forensic testing have been chosen partly because their alleles tend to be different in different people. For example, 2% of the population might have the alleles with 7 and 10 repeats at a particular STR locus; 1% might have the combination of 5 and 6; and so on. If we take a DNA molecule’s view of the population, human beings are containers for DNA and machines for copying and propagating them to the next generation of human beings. The different DNA molecules are swimming, so to speak, in a huge pool of humanity. All the possible alleles (the fives, sixes, sevens, and so on) form a large population, or pool, of alleles. Each allele constitutes a certain proportion of allele pool. Suppose, then, that a five-repeat allele represents 12% of all of the allele pool, a six-repeat allele contributes 20%, and so on, for all the alleles at a locus.
The first step in computing a random-match probability is to estimate these allele frequencies. Ideally, a probability sample from the human population of interest would be taken.74 We would start with a list of everyone who might have left the trace evidence, take a random sample of these people, and count the numbers of alleles of each length that are present in the sample. Unfortunately, a list of the people who comprise the entire population of possible suspects is almost never available; consequently, probability sampling from the directly relevant population is impossible. Probability sampling from a comparable population (with regard to the individuals’ DNA) is possible, but it is not the norm in studies of the distributions of genes in populations. Typically, convenience samples (from blood banks or paternity cases) are used.75 Rela-
74. Probability sampling is described in Kaye & Freedman, supra note 2, and Shari Seidman Diamond, Reference Guide on Survey Research, in this manual.
75. A few experts have testified that no meaningful conclusions can be drawn in the absence of random sampling. E.g., People v. Soto, 88 Cal. Rptr. 2d 34 (1999); State v. Anderson, 881 P.2d 29, 39 (N.M. 1994). The 1996 NRC report suggests that for the purpose of estimating allele frequencies, convenience sampling should give results comparable to random sampling, and it discusses procedures for estimating the random sampling error. NRC II, supra note 9, at 126–27, 146–48, 186. The courts
tively small samples can produce fairly accurate estimates of individual allele frequencies.76
Once the allele frequencies have been estimated, the next step in arriving at a random-match probability is to combine them. This requires some knowledge of how DNA is copied and recombined in the course of sexual reproduction and how human beings choose their mates.
2. The product rule for a randomly mating population
All scientists use simplified models of a complex reality. Physicists solve equations of motion in the absence of friction. Economists model exchanges among rational agents who bargain freely with no transaction costs. Population geneticists compute genotype frequencies in an infinite population of individuals who choose their mates independently of their alleles at the loci in question. Although geneticists describe this situation as random mating, geneticists know that people do not choose their mates by a lottery. “Random mating” simply indicates that the choices are uncorrelated with the specific alleles that make up the genotypes in question.
In a randomly mating population, the expected frequency of a pair of alleles at any single locus depends on whether the two alleles are distinct. If the offspring happens to inherit the same allele from each parent, the expected single-locus genotype frequency is the square of the allele frequency (p2). If a different allele is inherited from each parent, the expected single-locus genotype frequency is twice the product of the two individual allele frequencies (often written as 2p1p2).77 These proportions are known as Hardy-Weinberg proportions. Even if two populations with distinct allele frequencies are thrown together, within the limits of chance variation, random mating produces Hardy-Weinberg equilibrium in a single generation.
generally have rejected the argument that random samples are essential to valid or generally accepted random-match probabilities. See D.H. Kaye, Bible Reading: DNA Evidence in Arizona, 28 Ariz. St. L.J. 1035 (1996).
76. In the formative years of forensic DNA testing, defendants frequently contended that forensic databases were too small to give accurate estimates, but this argument generally proved unpersuasive. E.g., United States v. Shea, 957 F. Supp. 331, 341–43 (D.N.H. 1997); State v. Dishon, 687 A.2d 1074, 1090 (N.J. Super. Ct. App. Div. 1997); State v. Copeland, 922 P.2d 1304, 1321 (Wash. 1996). To the extent that the databases are comparable to random samples, confidence intervals are a standard method for indicating the uncertainty resulting from sample size. Unfortunately, the meaning of a confidence interval is subtle, and the estimate commonly is misconstrued. See Kaye & Freedman, supra note 2.
77. Suppose that 10% of the sperm in the gene pool of the population carry allele 1 (A1), and 50% carry allele 2 (A2). Similarly, 10% of the eggs carry A1, and 50% carry A2. (Other sperm and eggs carry other types.) With random mating, we expect 10% × 10% = 1% of all the fertilized eggs to be A1A1, and another 50% × 50% = 25% to be A2A2. These constitute two distinct homozygote profiles. Likewise, we expect 10% × 50% = 5% of the fertilized eggs to be A1A2 and another 50% × 10% = 5% to be A2A1. These two configurations produce indistinguishable profiles—a peak, band, or dot for A1 and another mark for A2. So the expected proportion of heterozygotes A1A2 is 5% + 5% = 10%.
Once the proportion of the population that has each of the single-locus genotypes for the forensic profile has been estimated, the proportion of the population that is expected to share the combination of them—the multilocus profile frequency—is given by multiplying all the single-locus proportions. This multiplication is exactly correct when the single-locus genotypes are statistically independent. In that case, the population is said to be in linkage equilibrium.
Early estimates of DNA genotype frequencies assumed that alleles were inherited independently within and across loci (Hardy-Weinberg and linkage equilibrium, respectively). Because the frequencies of the VNTR loci then in use were shown to vary across census groups (whites, blacks, Hispanics, Asians, and Native Americans), it became common to present the estimated genotype frequencies within each of these groups (in cases in which the “race” of the source of the trace evidence was unknown) or only in a particular census group (if the “race” of the source was known).78
3. The product rule for a structured population
Population geneticists understood that the equilibrium frequencies were only approximations and that the major racial populations are composed of ethnic subpopulations whose members tend to mate among themselves. Within each ethnic subpopulation, mating still can be random, but if, say, Italian Americans have allele frequencies that are markedly different than the average for all whites, and if Italian Americans only mate among themselves, then using the average frequencies for all whites in the basic product formula could understate—or overstate—a multilocus profile frequency for the subpopulation of Italian Americans. Similarly, using the population frequencies could understate—or overstate—the profile frequencies in the white population itself.
Consequently, if we want to know the frequency of an incriminating profile among Italian Americans, the basic product rule applied to the allele frequencies for whites in general could be in error; and there is even some chance that the rule will understate the profile frequency in the white population as a whole. Experts have disagreed, however, as to whether the major population groups are so severely structured that the departures from equilibrium would be substantial. Courts applying the Daubert and Frye rules for scientific evidence issued conflicting opinions as to the admissibility of basic product-rule estimates.79 A 1992 report from a committee of the National Academy of Sciences did not resolve the question, but a second committee concluded in 1996 that the basic product rule provided reasonable estimates in most cases, and it described a modified version of the product rule
78. The use of a range of estimates conditioned on race is defended, and several alternatives are discussed in Kaye, supra note 3, at 192–97; David H. Kaye, The Role of Race in DNA Evidence: What Experts Say, What California Courts Allow, 37 Sw. U. L. Rev. 303 (2008).
79. These legal and scientific developments are chronicled in detail in Kaye, supra note 3.
to account for population structure.80 By the mid-1990s, the population-structure objection to admitting random-match probabilities had lost its power.81
F. Probabilities, Probative Value, and Prejudice
Up to this point, we have described the random-match probabilities that commonly are presented in conjunction with the finding that the trace evidence sample contains DNA of the same type as the defendant’s. We have concentrated on the methods used to compute the probabilities. Assuming that these methods meet Daubert’s demand for scientific validity and reliability (or, in many states, Frye’s requirement of general acceptance in the scientific community) and thus satisfy Federal Rule of Evidence 702, a further issue can arise under Rule 403: To what extent will the presentation assist the jury in understanding the meaning of a match so that the jury can give the evidence the weight that it deserves? This question involves psychology and law, and we summarize the arguments about probative value and prejudice that have been made in litigation and in the legal and scientific literature. We take no position on how the legal issue of the admissibility of any particular statistic generally should be resolved under the balancing standard of Rule 403. The answer may turn not only on the general features of the evidence described here, but on the context and circumstances of particular cases.
1. Frequencies and match probabilities
a. Argument: Frequencies or robabilities are prejudicial because they are so small
The most common form of expert testimony about matching DNA involves an explanation of how the laboratory ascertained that the defendant’s DNA has the profile of the forensic sample plus an estimate of the profile frequency or random-match probability. It has been suggested, however, that jurors do not understand probabilities in general, and that infinitesimal match probabilities will so bedazzle jurors that they will not appreciate the other evidence in the case or any innocent explanations for the match.82 Empirical research into this hypothesis has been limited,83 and commentators have noted that remedies short of exclusion
80. The 1996 committee’s recommendations for computing random-match probabilities with broad populations and particular subpopulations are summarized in the previous edition of this guide. The 1992 committee had proposed a more conservative (and less elegant) method of dealing with variations across subpopulations (the “ceiling principle”), also described in the previous edition.
81. See, e.g., Kaye, supra note 3.
82. Cf. Gov’t of the Virgin Islands v. Byers, 941 F. Supp. 513, 527 (D.V.I. 1996) (“Vanishingly small probabilities of a random match may tend to establish guilt in the minds of jurors and are particularly suspect.”).
83. This research is tabulated in David H. Kaye et al., Statistics in the Jury Box: Do Jurors Understand Mitochondrial DNA Match Probabilities? 4 J. Empirical Legal Stud. 797 (2007). The findings do
are available.84 Thus, although there once was a line of cases that excluded probability testimony in criminal matters, by the mid-1990s, no jurisdiction excluded DNA match probabilities on this basis.85 The opposite argument—that relatively large random-match probabilities are prejudicial—also has been advanced without success.86
b. Argument: Frequencies or probabilities are prejudicial because they might be transposed
A related concern is that the jury will misconstrue the random-match probability as the probability that the evidence DNA came from a random individual.87 The words are almost identical, but the probabilities can be quite different. The random-match probability is the probability that the suspect has the DNA genotype of the crime scene sample if he is not the true source of that sample (and is unrelated to the true source). The tendency to invert or transpose the probability—to go from a one-in-a-million chance if the suspect is not the source to a million-to-one chance that the suspect is the source is known as the fallacy of the transposed conditional.88 To appreciate that the transposition is fallacious, consider the probability
not clearly support the argument that jurors will overweight the probability, but the details of how the probability is presented and countered may be important.
84. According to the 1996 NRC committee, suitable cross-examination, defense experts, and jury instructions might reduce the risk that small estimates of the match probability will produce an unwarranted sense of certainty and lead a jury to disregard other evidence. NRC II, supra note 9, at 197.
85. E.g., United States v. Chischilly, 30 F.3d 1144 (9th Cir. 1994) (citing cases); State v. Weeks, 891 P.2d 477, 489 (Mont. 1995) (rejecting the argument that “the exaggerated opinion of the accuracy of DNA testing is prejudicial, as juries would give undue weight and deference to the statistical evidence” and “that the probability aspect of the DNA analysis invades the province of the jury to decide the guilt or innocence of the defendant”).
86. See United States v. Morrow, 374 F. Supp. 2d 51, 65 (D.D.C. 2005) (rejecting the argument because “the DNA evidence remains probative, and helps to corroborate other evidence and support the Government’s case as to the identity of the relevant perpetrators. Indeed, the low statistical significance actually benefits Defendants, as Defendants can argue that having random match probabilities running between 1:12 and 1:1 means that hundreds, if not thousands, of others in the Washington, D.C. area cannot be excluded as possible contributors as well.”).
87. Numerous opinions or experts present the random-match probability in this manner. E.g., State v. Davolt, 84 P.3d 456, 475 (Ariz. 2004) (stating that “the chance the saliva found on cigarette remains in the house did not belong to [the defendant] was one in 280 quadrillion for the Caucasian population”); Kaye et al., supra note 1, § 14.1.2(a) (collecting opinions reflecting this fallacy).
88. The transposition fallacy also is called the “prosecutor’s fallacy” in the legal literature—despite the fact that it hardly is limited to prosecutors. Our description of the fallacy is imprecise. In this context, the random-match probability is the chance that (A) the suspect has the crime scene genotype given that (B) he is not the true source. The probability that the match is random is the probability that (B) the individual tested has been selected at random given that (A) the individual has the requisite genotype. In general, for two events A and B, the probability of A given B, which we can write as P(A given B), does not equal P(B given A). See Kaye & Freedman, supra note 2. The claim that the probabilities are necessarily equal is the transposition fallacy. Id. (also noting instances of the fallacy in other types of litigation).
that a lawyer picked at random from all lawyers in the United States is an appellate judge. This “random-judge probability” is practically zero. But the probability that a person randomly selected from the current appellate judiciary is a lawyer is one. The random-judge probability, P(judge given lawyer), does not equal the transposed probability P(lawyer given judge). Likewise, the random-match probability P(genotype given unrelated source) does not necessarily equal P(unrelated source given genotype).89
No federal court has excluded a random-match probability (or, for that matter, an estimate of the small frequency of a DNA profile in the general population) as unfairly prejudicial simply because the jury might misinterpret it as a probability that the defendant is the source of the forensic DNA.90 Courts, however, have noted the need to have the concept “properly explained,”91 and prosecutorial or expert misrepresentations of the random-match probabilities for
89. To avoid this fallacious reasoning by jurors, some scientific and legal commentators have urged the exclusion of random-match probabilities. In response, the 1996 NRC committee suggested that “if the initial presentation of the probability figure, cross-examination, and opposing testimony all fail to clarify the point, the judge can counter [the fallacy] by appropriate instructions to the jurors that minimize the possibility of cognitive errors.” NRC II, supra note 9, at 198 (footnote omitted). The committee suggested the following instruction to define the random-match probability:
In evaluating the expert testimony on the DNA evidence, you were presented with a number indicating the probability that another individual drawn at random from the [specify] population would coincidentally have the same DNA profile as the [bloodstain, semen stain, etc.]. That number, which assumes that no sample mishandling or laboratory error occurred, indicates how distinctive the DNA profile is. It does not by itself tell you the probability that the defendant is innocent.
Id. at 198 n.93. An alternative adopted in England is to confine the prosecution to stating a frequency rather than a probability. See Kaye et al., supra note 1, § 14.1.2(b); cf. D.H. Kaye, The Admissibility of “Probability Evidence” in Criminal Trials—Part II, 27 Jurimetrics J. 160, 168 (1987) (similar proposal).
The NRC committee also noted the opposing “defendant’s fallacy” of dismissing or undervaluing the matches with high likelihood ratios because other matches are to be expected in unrealistically large populations of potential suspects. For example, defense counsel might argue that (1) with a random-match probability of one in a million, we would expect to find three or four unrelated people with the requisite genotypes in a major metropolitan area with a population of 3.6 million; (2) the defendant just happens to be one of these three or four, which means that the chances are at least 2 out of 3 that someone unrelated to the defendant is the source; so (3) the DNA evidence does nothing to incriminate the defendant. The problem with this argument is that in a case involving both DNA and non-DNA evidence against the defendant, it is unrealistic to assume that there are 3.6 million equally likely suspects. When juries are confronted with both fallacies, the defendant’s fallacy seems to dominate. NRC II, supra note 9, at 198; cf. Jonathan J. Koehler, The Psychology of Numbers in the Courtroom: How to Make DNA-Match Statistics Seem Impressive or Insufficient, 74 S. Cal. L. Rev. 1275 (2001) (discussing ways of framing the evidence that make it more or less persuasive).
90. See, e.g., United States v. Morrow, 374 F. Supp. 2d 51, 66 (D.D.C. 2005) (“careful oversight by the district court and proper explanation can easily thwart this issue”).
91. United States v. Shea, 957 F. Supp. 331, 345 (D.N.H. 1997); see also United States v. Chischilly, 30 F.3d 1144, 1158 (9th Cir. 1994) (stating that the government must be “careful to frame the DNA profiling statistics presented at trial as the probability of a random match, not the probability of the defendant’s innocence that is the crux of the prosecutor’s fallacy”).
DNA and other trace evidence have produced reversals or contributed to the setting aside of verdicts.92
c. Argument: Random-match probabilities that are smaller than false-positive error probabilities are irrelevant or prejudicial
Some scientists and lawyers have maintained that match probabilities are logically irrelevant when they are far smaller than the probability of a frameup, a blunder in labeling samples, cross-contamination, or other events that would yield a false positive.93 The argument is that the jury should concern itself only with the chance that the forensic sample is reported to match the defendant’s profile even though the defendant is not the source. Match probabilities do not express this chance unless the probability of a false-positive report (because of fraud or an error in the collection, handling, or analysis of the DNA samples) is essentially zero. The mathematical observation has led to the argument that because these other possible explanations for a match are more probable than the very small random-match probabilities for most STR profiles, the latter probabilities are irrelevant. Commentators have crafted theoretical, doctrinal, and practical rejoinders to this claim.94 The essence of the counterargument is that it is logical to give jurors information about kinship or random-match probabilities because, even if these numbers do not give the whole picture, they address pertinent hypotheses about the true source of the trace evidence.
It also has been argued that even if very small match probabilities are logically relevant, they are unfairly prejudicial in that they will cause jurors to neglect the probability of a match arising due to a false-positive laboratory error.95 A court
92. E.g., United States v. Massey, 594 F.2d 676, 681 (8th Cir. 1979) (explaining that in closing argument about hair evidence, “the prosecutor ‘confuse[d] the probability of concurrence of the identifying marks with the probability of mistaken identification’”) (alteration in original). The Supreme Court noted the transposition fallacy in the prosecution’s presentation of DNA evidence as a basis for a federal writ of habeas corpus in McDaniel v. Brown, 130 S. Ct. 665 (2010) (per curiam). The Court unanimously held that the prisoner had not properly raised the issue of whether this error amounted to a violation of due process. For comments on that issue, see Kaye, supra note 71.
93. E.g., Jonathan J. Koehler et al., The Random Match Probability in DNA Evidence: Irrelevant and Prejudicial? 35 Jurimetrics J. 201 (1995); Richard C. Lewontin & Daniel L. Hartl, Population Genetics in Forensic DNA Typing, 254 Science 1745, 1749 (1991) (“[p]robability estimates like 1 in 738,000,000,000,000…are terribly misleading because the rate of laboratory error is not taken into account”).
94. See Kaye et al., supra note 1, § 14.1.1 (discussing the issue).
95. Some commentators believe that this prejudice is so likely and so serious that “jurors ordinarily should receive only the laboratory’s false positive rate….” Richard Lempert, Some Caveats Concerning DNA as Criminal Identification Evidence: With Thanks to the Reverend Bayes, 13 Cardozo L. Rev. 303, 325 (1991) (emphasis added). The 1996 NRC committee was skeptical of this view, especially when the defendant has had a meaningful opportunity to retest the DNA at a laboratory of his or her choice, and it suggested that judicial instructions can be crafted to avoid this form of prejudice. NRC II, supra note 9, at 199. Pertinent psychological research includes Dale A. Nance & Scott B. Morris, Juror Understanding of DNA Evidence: An Empirical Assessment of Presentation Formats for Trace
that shares this concern might require the expert who presents a random-match probability also to report a probability that the laboratory is mistaken about the profiles. Of course, for reasons given in Section III.B.2, some experts would deny that they can provide a meaningful statistic for the case at hand, but it has been pointed out that they could report the results of proficiency tests and leave it to the jury to use this figure as best it can in considering whether a false-positive error has occurred.96 In any event, the courts have been unreceptive to efforts to replace random-match probabilities with a blended figure that incorporates the risk of a false-positive error97 or to exclude random-match probabilities that are not accompanied by a separate false-positive error probability.98
Evidence with a Relatively Small Random Match Probability, 34 J. Legal Stud. 395 (2005); Dale A. Nance & Scott B. Morris, An Empirical Assessment of Presentation Formats for Trace Evidence with a Relatively Large and Quantifiable Random Match Probability, 42 Jurimetrics J. 1 (2002); Jason Schklar & Shari Seidman Diamond, Juror Reactions to DNA Evidence: Errors and Expectancies, 23 Law & Hum. Behav. 159, 179 (1999) (concluding that separate figures for laboratory error and a random match to a correctly ascertained profile are desirable in that “[j]urors…may need to know the disaggregated elements that influence the aggregated estimate as well as how they were combined in order to evaluate the DNA test results in the context of their background beliefs and the other evidence introduced at trial”).
96. Cf. Williams v. State, 679 A.2d 1106, 1120 (Md. 1996) (reversing because the trial court restricted cross-examination about the results of proficiency tests involving other DNA analysts at the same laboratory). But see United States v. Shea, 957 F. Supp. 331, 344 n.42 (D.N.H. 1997) (“The parties assume that error rate information is admissible at trial. This assumption may well be incorrect. Even though a laboratory or industry error rate may be logically relevant, a strong argument can be made that such evidence is barred by Fed. R. Evid. 404 because it is inadmissible propensity evidence.”).
97. United States v. Ewell, 252 F. Supp. 2d 104, 113–14 (D.N.J. 2003) (stating that exclusion of the random-match probability is not justified when “the defendant’s argument is not based on evidence of actual errors by the laboratory, but instead has simply challenged the Government’s failure to quantify the rate of laboratory error,” while “the Government has demonstrated the scientific method has a virtually zero rate of error, and that it employs sufficient procedures and controls to limit laboratory error,” and the defendant had an expert who could testify to the probability of error); United States v. Shea, 957 F. Supp. 331, 334–45 (D.N.H. 1997) (holding that separate figures for match and error probabilities are not prejudicial); People v. Reeves, 109 Cal. Rptr. 2d 728, 753 (Ct. App. 2001) (holding that probability of laboratory error need not be combined with random-match probability); Armstead v. State, 673 A.2d 221, 245 (Md. 1996) (finding that the failure to combine a random-match probability with an error rate on proficiency tests that was many orders of magnitude greater (and that was placed before the jury) did not deprive the defendant of due process); State v. Tester, 968 A.2d 895 (Vt. 2009).
98. United States v. Trala, 162 F. Supp. 2d 336, 350–51 (D. Del. 2001) (stating that presenting a nonzero laboratory error rate is not a condition of admissibility, and Daubert does not require separate figures for match and error probabilities to be combined); United States v. Lowe, 954 F. Supp. 401, 415–16 (D. Mass. 1997), aff’d, 145 F.3d 45 (1st Cir. 1998) (finding that a “theoretical” error rate need not be presented when quality assurance standards have been followed and defendant had the opportunity to retest the sample); Roberts v. United States, 916 A.2d 922, 930–31 (D.C. 2007) (finding that presenting a laboratory error rate is not a condition of admissibility); Roberson v. State, 16 S.W.3d 156, 168 (Tex. Crim. App. 2000) (finding that error rate not needed when laboratory was accredited and underwent blind proficiency testing); Tester, 968 A.2d 895 (stating that when the laboratory chemist stated that “[t]here is no error rate to report” because the number of proficiency
Sufficiently small probabilities of a match for close relatives and unrelated members of the suspect population undermine the hypotheses of kinship and coincidence. Adequate safeguards and checks for possible laboratory error make that explanation of the finding of matching genotypes implausible. The inference that the defendant is the source of the crime scene DNA is then secure. But this mode of reasoning by elimination is not the only way to analyze DNA evidence. This subsection and the next describe alternatives—likelihoods and posterior probabilities—that some statisticians prefer and that have been used in a growing number of court cases.
To choose between two competing hypotheses, one can compare how probable the evidence is under each hypothesis. Suppose that the probability of a match in a well-run laboratory is close to 1 when the samples both contain only the defendant’s DNA, while both the probability of a coincidental match and the probability of a match to a close relative are close to 0. In these circumstances, the DNA profiling strongly supports the claim that the defendant is the source, because the observed outcome—the match—is many times more probable when the defendant is the source than when someone else is. How many times more probable? Suppose that there is a 1% chance that the laboratory would miss a true match, so that the probability of its finding a match when the defendant is the source is 0.99. Suppose further that p = 0.00001 is the random-match probability. Then the match is 0.99/0.00001, or 99,000 times more likely to be seen if the defendant is the source than if an unrelated individual is. Such a ratio is called a likelihood ratio, and a likelihood ratio of 99,000 means that the DNA profiling supports the claim of identity 99,000 times more strongly than it supports the hypothesis of coincidence.99
Likelihood ratios have been presented in court in many cases. They are routinely introduced under the name “paternity index” in civil and criminal cases that involve DNA testing for paternity.100 Experts also have used them in cases in which the issue is whether two samples originated from the same individual. For example, in one California case, an expert stated that “for the Caucasian population, the evidence DNA profile was approximately 1.9 trillion times more likely to match appellant’s DNA profile if he was the contributor of that DNA rather than some unknown, unrelated individual; for the Hispanic population, it was 2.6 trillion times more likely; and for the African-American population, it was about 9.1 trillion times more likely.”101 And, as explained below (Section V.C), likeli-
trials was insufficient, the random-match probability was admissible and preferable to presenting the finding of a match with no accompanying statistic).
99. Another likelihood ratio would give the relative likelihood of the hypotheses of identity and a falsely declared match arising from an error in the laboratory. See supra Section IV.F.1.
100. See Kaye, supra note 3; 1 McCormick on Evidence, supra note 3, § 211.
101. People v. Prince, 36 Cal. Rptr. 3d 300, 310 (Ct. App. 2005), review denied, 142 P.3d 1184 (Cal. 2006).
hood ratios are especially useful for samples that are mixtures of DNA from several people.
The major objection to likelihoods is not statistical, but psychological.102 As with random-match probabilities, they are easily transposed.103 With random-match probabilities, we saw that courts have reasoned that the possibility of transposition does not justify a blanket rule of exclusion. The same issue has not been addressed directly for likelihood ratios.
The likelihood ratio expresses the relative strength of two hypotheses, but the judge or jury ultimately must assess a different type of quantity—the probability of the hypotheses themselves. An elementary rule of probability theory known as Bayes’ theorem yields this probability. The theorem states that the odds in light of the data (here, the observed profiles) are the odds as they were known prior to receiving the data times the likelihood ratio. More succinctly, posterior odds = likelihood ratio × prior odds.104 For example, if the relevant match probability105 were 1/100,000, and if the chance that the laboratory would report a match between samples from the same source were 0.99, then the likelihood ratio would be 99,000, and the jury could be told how the DNA evidence raises various prior probabilities that the defendant’s DNA is in the evidence sample.106
102. For legal commentary and additional cases upholding the admission of likelihood ratios over objections based on Frye and Daubert, see Kaye et al., supra note 1, § 14.2.2.
103. United States v. Thomas, 43 M.J. 626 (A.F. Ct. Crim. App. 1995), provides an example. In this murder case, a military court described testimony from a population geneticist that “conservatively, it was 76.5 times more likely that the samples…came from the victim than from someone else in the Filipino population.” Id. at 635. Yet, this is not what the DNA testing showed. A more defensible statement is that “the match between the bloodstains was 76.5 times more probable if the stains came from the victim than from an unrelated Filipino” or “the match supports the hypothesis that the stains came from the victim 76.5 times more than it supports the hypothesis that they came from an unrelated Filipino woman.” Kaye et al., supra note 7, § 14.2.2.
104. Odds and probabilities are two ways to express chances quantitatively. If the probability of an event is P, the odds are P/(1 − P). If the odds are O, the probability is O/(O + 1). For instance, if the probability of rain is 2/3, the odds of rain are 2 to 1 because (2/3)/(1 − 2/3) = (2/3)/(1/3) = 2. If the odds of rain are 2 to 1, then the probability is 2/(2 + 1) = 2/3.
105. By “relevant match probability,” we mean the probability of a match given a specified type of kinship or the probability of a random match in the relevant suspect population. For relatives more distantly related than second cousins, the probability of a chance match is nearly as small as for persons of the same subpopulation. Devlin & Roeder, supra note 63, § 18-3.1.3, at 724.
106. If this procedure is followed, the analyst could explain that these calculations rest on many premises, including the premise that the genotypes have been correctly determined. See, e.g., Richard Lempert, The Honest Scientist’s Guide to DNA Evidence, 96 Genetica 119 (1995). If the jury accepted these premises and also decided to accept the hypothesis of identity over those of kinship and coincidence, it still would be open to the defendant to offer explanations of how the forensic samples came to include his or her DNA even though he or she is innocent.
One difficulty with this use of Bayes’ theorem is that the computations consider only one alternative to the claim of identity at a time. As indicated earlier, however, several rival hypotheses might apply in a given case. If the DNA in the crime scene sample is not the defendant’s, is it from his father, his brother, his uncle, or another relative? Is the true source a member of the same subpopulation? Or is the source a member of a different subpopulation in the same general population? In principle, the likelihood ratio can be generalized to a likelihood function that takes on suitable values for every person in the world, and the prior probability for each person can be cranked into a general version of Bayes’ rule to yield the posterior probability that the defendant is the source. In this vein, some commentators suggest that Bayes’ rule be used to combine the various likelihood ratios for all possible degrees of kinship and subpopulations.107
As with likelihood ratios, Bayes’ rule is routine in cases involving parentage testing. Some courts have held that the “probability of paternity” derived from the formula is inadmissible in criminal cases, but most have reached the opposite conclusion, at least when the prior odds used in the calculation are disclosed to the jury.108 An extended literature has grown up on the subject of how posterior probabilities might be useful in criminal cases.109
G. Verbal Expressions of Probative Value
Having surveyed the issues related to the value and dangers of probabilities and statistics for DNA evidence, we turn to a related issue that can arise under Rules 702 and 403: Should an expert be permitted to offer a nonnumerical judgment about the DNA profiles? Many courts have held that a DNA match is inadmissible unless the expert attaches a scientifically valid number to the match. Indeed, some opinions state that this requirement flows from the nature of science itself. However, this view has been challenged,110 and not all courts agree that an expert must explain the power of a DNA match in purely numerical terms.
107. David J. Balding, Weight-of-Evidence for Forensic DNA Profiles (2005); David J. Balding & Peter Donnelly, Inference in Forensic Identification, 158 J. Royal Stat. Soc’y Ser. A 21 (1995); cf. Lempert, supra note 69, at 458 (describing a similar procedure).
108. Kaye et al., supra note 1, § 14.3.2.
109. See id.; 1 McCormick on Evidence, supra note 3, § 211; David H. Kaye, Rounding Up the Usual Suspects: A Legal and Logical Analysis of DNA Database Trawls, 87 N.C. L. Rev. 425 (2009) (defending a Bayesian presentation by a defendant identified by a “cold hit” in a DNA database).
110. See, e.g., Commonwealth v. Crews, 640 A.2d 395, 402 (Pa. 1994) (explaining that “[t]he factual evidence of the physical testing of the DNA samples and the matching alleles, even without statistical conclusions, tended to make appellant’s presence more likely than it would have been without the evidence, and was therefore relevant.”). The 1996 NRC committee wrote that science only demands “underlying data that permit some reasonable estimate of how rare the matching characteristics actually are,” and “[o]nce science has established that a methodology has some individualizing power, the legal system must determine whether and how best to import that technology into the trial process.” NRC II, supra note 9, at 192.
1. “Rarity” or “strength” testimony
Instead of presenting numerical frequencies or match probabilities, a scientist could characterize a 13-locus STR profile as “rare,” “extremely rare,” or the like. Instead of quoting a numerical likelihood ratio, the analyst could refer to the match as “powerful,” “very strong evidence,” and so on. At least one state supreme court has endorsed this qualitative approach as a substitute to the presentation of quantitative estimates.111
2. Source or uniqueness testimony
The most extreme case of a purely verbal description of the infrequency of a profile occurs when that profile can be said to be unique. Of course, the uniqueness of any object, from a snowflake to a fingerprint, in a population that cannot be enumerated never can be proved directly. As with all sample evidence, one must generalize from the sample to the entire population. There is always some probability that a census would prove the generalization to be false. Over a decade ago, the second NRC committee therefore wrote that “[t]here is no ‘bright-line’ standard in law or science that can pick out exactly how small the probability of the existence of a given profile in more than one member of a population must be before assertions of uniqueness are justified…. There might already be cases in which it is defensible for an expert to assert that, assuming that there has been no sample mishandling or laboratory error, the profile’s probable uniqueness means that the two DNA samples come from the same person.”112 Before concluding that a DNA profile is unique in a given population, however, a careful expert also should consider not only the random-match probability (which pertains to unrelated individuals) but also the chance of a match to a close relative. Indeed, the possible existence of an unknown, identical twin also means that a scientist never can be absolutely certain that crime scene evidence could have come from only the defendant.
Courts have accepted or approved of expert assertions of uniqueness or of individual source identification.113 For these assertions to be justified, a large
111. State v. Bloom, 516 N.W.2d 159, 166–67 (Minn. 1994) (“Since it may be pointless to expect ever to reach a consensus on how to estimate, with any degree of precision, the probability of a random match and that, given the great difficulty in educating the jury as to precisely what that figure means and does not mean, it might make sense to simply try to arrive at a fair way of explaining the significance of the match in a verbal, qualitative, nonquantitative, nonstatistical way.”). A related question is whether an expert should be allowed to declare a match without adding any information on how common or rare the profile is. For discussion of such pure “defendant-not-excluded” testimony, see United States v. Morrow, 374 F. Supp. 2d 51 (D.D.C. 2005); Kaye et al., supra note 1, § 15.4.
112. NRC II, supra note 9, at 194.
113. E.g., United States v. Davis, 602 F. Supp. 2d 658 (D. Md. 2009) (“the random match probability figures…are sufficiently low so that the profile can be considered unique”); People v. Baylor, 118 Cal. Rptr. 2d 518, 522 (Ct. App. 2002) (testimony that “defendant had a unique DNA
number of sufficiently polymorphic loci must have been tested, making the probabilities of matches to both relatives and unrelated individuals so tiny that the probability of finding another person who could be the source within the relevant population is negligible.114
V. Special Issues in Human DNA Testing
Mitochondria are small structures, with their own membranes, found inside the cell but outside its nucleus. Inside these organelles, molecules are broken down to supply energy. Mitochondria have a small genome—a circle of 16,569 nucleotide base pairs within the mitochondrion—that bears no relation to the comparatively monstrous chromosomal genome in the cell nucleus.115
Mitochondrial DNA (mtDNA) has four features that make it useful for forensic DNA testing. First, the typical cell, which has but one nucleus, contains hundreds or thousands of nearly identical mitochondria. Hence, for every copy of
profile that ‘probably does not exist in anyone else in the world.’”); State v. Hauge, 79 P.3d 131 (Haw. 2003) (uniqueness); Young v. State, 879 A.2d 44, 46 (Md. 2005) (holding that “when a DNA method analyzes genetic markers at sufficient locations to arrive at an infinitesimal random match probability, expert opinion testimony of a match and of the source of the DNA evidence is admissible”; hence, it was permissible to introduce a report providing no statistics but stating that “(in the absence of an identical twin), Anthony Young (K1) is the source of the DNA obtained from the sperm fraction of the Anal Swab (R1).”); State v. Buckner, 941 P.2d 667, 668 (Wash. 1997) (finding that in light of 1996 NRC Report, “we now conclude there should be no bar to an expert giving his or her expert opinion that, based upon an exceedingly small probability of a defendant’s DNA profile matching that of another in a random human population, the profile is unique.”).
114. We apologize for the length of this sentence, but there are three distinct probabilities that arise in speaking of the uniqueness of DNA profiles. First, there is the probability of a match to a single, randomly selected individual in the population. This is the random-match probability. Second, there is the probability that the particular profile is unique. This probability involves pairing the profile with every member of the population. Third, there is the probability that all pairs of all profiles are unique. The first probability is larger than the second, which is many times larger than the third. Uniqueness or source testimony need only establish that the one DNA profile in the trace evidence is unique—and not that all DNA profiles are unique. Thus, it is the second probability, properly computed, that must be quite small to warrant the conclusion that no one but the defendant (and any identical twins) could be the source of the crime scene DNA. See David H. Kaye, Identification, Individuality, and Uniqueness: What’s the Difference? 8 Law, Probability & Risk 85 (2009).
Formulas for estimating all these probabilities are given in NRC II, supra note 9, but DNA analysts and judges sometimes infer uniqueness on the basis of incorrect intuitions about the size of the random-match probability. See Balding, supra note 107, at 148 (2005) (describing “the uniqueness fallacy”); cf. State v. Lee, 976 So. 2d 109, 117 (La. 2008) (incorrect but harmless miscalculation).
115. Mitochondria probably started out as bacteria that were engulfed by cells eons ago. Some of their genes have migrated to the chromosomes, but STR and other DNA sequences in the nucleus are not physically or statistically associated with the sequences of the DNA in the mitochondria.
chromosomal DNA, there are hundreds or thousands of copies of mitochondrial DNA. This means that it is possible to detect mtDNA in samples, such as bone and hair shafts, that contain too little nuclear DNA for conventional typing.
Second, two “hypervariable” regions that tend to be different in different individuals lie within the “control region” or “D-loop” (displacement loop) of the mitochondrial genome.116 These regions extend for a bit more than 300 base pairs each—short enough to be typable even in highly degraded samples such as very old human remains.
Third, mtDNA comes solely from the egg cell.117 For this reason, mtDNA is inherited maternally, with no fatherly contribution:118 Siblings, maternal half-siblings, and others related through maternal lineage normally possess the same mtDNA sequence. This feature makes mtDNA particularly useful for associating persons related through their maternal lineage. It has been exploited to identify the remains of the last Russian tsar and other members of the royal family, of soldiers missing in action, and of victims of mass disasters.
Finally, point mutations accumulate in the noncoding D-loop without altering how the mitochondrion functions. Hence, a single individual can develop distinct internal populations of mitochondria.119 As discussed below, this phenomenon, known as heteroplasmy, complicates the interpretation of mtDNA sequences. Yet, it is mutations that make mtDNA polymorphic and hence useful in identifying individuals. Over time, mutations in egg cells can propagate to later generations, producing more heterogeneity in mitochondrial genomes in the human population.120 This polymorphism allows scientists to compare mtDNA from crime scenes to mtDNA from given individuals to ascertain whether the tested individuals are within the maternal line (or another coincidentally matching maternal line) of people who could have been the source of the trace evidence.
The small mitochondrial genome can be analyzed with a PCR-based method that gives the order of all the base pairs.121 The sequences of two samples—say, DNA extracted from a hair shaft found at a crime scene and hairs plucked from a suspect—then can be compared. Most analysts describe the results in terms on
116. A third, somewhat less polymorphic, region in the D-loop can be used for additional discrimination. The remainder of the control region, although noncoding, consists of DNA sequences that are involved in the transcription of the mitochondrial genes. These control sequences are essentially the same in everyone (monomorphic).
117. The relatively few mitochondria in the spermatozoan that fertilizes the egg cell soon degrade and are not replicated in the multiplying cells of the pre-embryo.
118. The possibility of paternal contributions to mtDNA in humans is discussed in, e.g., John Buckleton et al., Nonautosomal Forensic Markers, in Forensic DNA Evidence Interpretation 299, 302 (John Buckleton et al. eds., 2005).
119. A single tissue has only one mitotype; another tissue from the same individual might have another mitotype; a third might have both mitotypes.
120. Evolutionary studies suggest an average mutation rate for the mtDNA control region of as little as one nucleotide difference every 300 generations, or one difference every 6000 years.
121. Other methods to ascertain the base-pair sequences also are available.
inclusions and exclusions, although, in principle, a likelihood ratio is better suited to cases in which there are slight sequence differences.122 In the simplest case, the two sequences show a good number of differences (a clear exclusion), or they are identical (an inclusion). In such cases, mitotyping can exclude individuals as the source of stray hairs even when the hairs are microscopically indistinguishable.123
As with nuclear DNA, to indicate the significance of the match, analysts usually estimate the frequency of the sequence in some population. The estimation procedure is actually much simpler with mtDNA. It is not necessary to combine any allele frequencies because the entire mtDNA sequence, whatever its internal structure may be, is inherited as a single unit (a “haplotype”). In other words, the sequence itself is like a single allele, and one can simply see how often it occurs in a sample of unrelated people.124
Laboratories therefore refer to databases of mtDNA sequences to see how often the type in question has been seen before. Often, the mtDNA sequence from the crime scene is not represented in the database, indicating that it is a relatively rare sequence. For example, in State v. Pappas,125 the reference database consisted of 1219 mtDNA sequences from whites, and it did not include the sequence that was present in the hairs near the crime scene and in the defendant. Thus, this particular sequence was observed once (at the crime scene) out of 1220 times (adding the new sequence to the 1219 different sequences on file). This would correspond to a population frequency of 0.082%. However, to account for sampling error (the inevitable differences between random samples and the population from which they are drawn), a laboratory might use a slightly different estimate. In general, laboratories count the occurrences in the database and take the upper end of a 95% confidence interval around the corresponding proportion.126 Applying this logic, an FBI analyst in Pappas testified to “the maximum match probability…of three in 1000…. [O]ut of 1000 randomly selected persons, it could be expected that three persons would share the same mtDNA type as the defendant.”127 The basic idea is that even if 3/1000 people in the white population have the sequence, there still is a 5% chance that it would not show up in a specific (randomly drawn) database of size 1219; hence, 3/1000 is a reasonable
122. The likelihood-ratio approach is developed in Buckleton et al., supra note 118.
123. The implications of this fact for the admissibility of microscopic hair analysis is discussed in Kaye, supra note 3.
124. In this context, “unrelated people” means individuals with a different maternal lineage.
125. 776 A.2d 1091 (Conn. 2001).
126. The Reference Manual on Statistics discusses the meaning of a confidence interval. It has been argued that instead of using x/N in forming the confidence interval, one should use the proportion (x + 1)/(N + 1), where x is the number of matching sequences in the database and N is the size of the database. After all, following the testing, x + 1 is the number of times that the sequence has been seen in N + 1 individuals. This is the reasoning that produced the point estimate of 1/1220 rather than 0/1219. For large databases, this alteration will make little difference in the confidence interval.
127. 776 A.2d at 1111 (emphasis added).
upper estimate for the population frequency.128 If the population frequency of the sequence in unrelated whites were much larger, the chance that the sequence would have been missed in the database sampling would be even less than 5%.
Computations that rely on databases of major population groups (such as whites) assume that the reference database is a representative sample of a population of unrelated individuals who might have committed the alleged crime. This assumption is justified if there has been sufficient random mating within the racial population. In principle, the adjustment that accounts for population structure (see supra Section IV.E.3) could be used, but how large the adjustment should be is not clear.129 Statistics derived from many databases from different locations also have been proposed.130 An alternative is to develop local databases that would reflect the proportion of all the people in the vicinity of the crime possessing each possible mitotype.131 Until these databases exist, an expert might give rather restricted quantitative testimony. In Pappas, for example, the expert could have said that the hairs and the defendants have the same mitotype and that this mitotype did not appear in a group of 1219 other people in a national sample, and the expert could have refrained from offering any estimate of the frequency in all whites. This restricted presentation suggests that the match has some probative value, but a court might need to consider whether it is sufficient to leave it to the jury to decide how to weigh the fact of the match and the absence of the same sequence in a convenience sample that might—or might not—be representative of the local white population.
Another issue is heteroplasmy. The simple inclusion-exclusion approach must be modified to account for the fact that the same individual can have detectably different mitotypes in different tissues or even in different cells in the same tissue. To understand the implications of heteroplasmy, we need to understand how it comes into existence.132 Heteroplasmy can occur because of mtDNA mutations during the division of adult cells, such as those at the roots of hair shafts. These new mitotypes are confined to the individual. They will not be passed on to future generations. Heteroplasmy also can result from a mutation contained in the egg cell that grew into an individual. Such mutations can make their way into succeeding generations, establishing new mitotypes in the population. But this is
128. In general, if the sequence does not exist in the database of size N, the upper 95% confidence limit is approximately 3/N. E.g., J.A. Hanley & A. Lipp-Hand, If Nothing Goes Wrong, Is Everything All Right? Interpreting Zero Numerators, 249 JAMA 1743 (1983). In Pappas, 3/N is 3/1219 = 0.25%, which rounds off to the 3 per 1000 figure quoted by the FBI analyst.
129. See Buckleton et al., supra note 118.
130. T. Egeland & A. Salas, Statistical Evaluation of Haploid Genetic Evidence, 1 Open Forensic Sci. J. 4 (2008).
131. Id.; see also F.A. Kaestle et al., Database Limitations on the Evidentiary Value of Forensic Mitochondrial DNA Evidence, 43 Am. Crim. L. Rev. 53 (2006).
132. An entertaining discussion can be found in Brian Sykes, The Seven Daughters of Eve: The Science That Reveals Our Genetic Ancestry 55–57, 62, 77–78 (2001).
an uncertain process. Eggs cells contain many mitochondria, and the mature egg cell will not contain just the mutation—it will house a mixed population of the old-style mitochondria and a number of the mutated ones (with DNA that usually differs from the original at a single base pair). Figuratively speaking, the original mtDNA sequence and the mutated version fight it out for several generations until one of them becomes “fixed” in the population. In the interim, the progeny of the mutated egg cell will harbor both strains of mitochondria.
When mtDNA from a crime scene sample is compared to a suspect’s sample, there are three possibilities: (1) neither sample is detectably heteroplasmic; (2) one sample displays heteroplasmy, but the other does not; (3) both samples display heteroplasmy. In each scenario, the comparison can produce an exclusion or an inclusion:
- Neither sample heteroplasmic. In the first situation, if the sequence in the crime scene sample is markedly different from the sequence in the suspect’s sample, then the suspect is excluded. But heteroplasmy could be the reason for a difference of only a single base or so. For example, the sequence in a hair shaft coming from the suspect could be a slight mutation of the dominant sequence in the suspect. Therefore, the FBI treats a difference at a single base pair as inconclusive.133 When the one mtDNA sequence characteristic of each sample is identical, the issue becomes how to use the reference database of mtDNA sequences, as discussed above.
- Suspect’s sample heteroplasmic, crime scene sample not. One version of the second scenario arises when heteroplasmy is seen in the suspect’s tissues but not in the crime scene sample. If the crime scene sequence is not close to either of the suspect’s sequences, then the suspect is excluded. If it is identical to one of the suspect’s sequences, then the suspect is included, and a suitable reference database should indicate how infrequent such an inclusion would be. If crime scene DNA is one base pair removed from either of the suspect’s sequences, then the result is inconclusive.
133. Scientific Working Group on DNA Analysis Methods (SWGDAM), Guidelines for Mitochondrial DNA (mtDNA) Nucleotide Sequence Interpretation, Forensic Sci. Comm., Apr. 2003, available at http://www.fbi.gov/hq/lab/fsc/backissu/april2003/swgdammitodna.htm. But see Vaughn v. State, 646 S.E.2d 212, 215 (Ga. 2007) (apparently transforming the statement that a suspect “cannot be excluded” when “there is a single base pair difference” into “a match”). These inconclusive sequences contribute to the number of people who would not be excluded. Therefore, in Pappas, it is misleading to conclude “that approximately 99.75% of the Caucasian population could be excluded as the source of the mtDNA in the sample.” 776 A.2d 1091, 1104 (Conn. 2001) (footnote omitted). This percentage neglects the individuals whose mtDNA sequences are off by one base pair. Along with the 0.25% who are included because their mtDNA matches completely, these one-off people would not be excluded. An analyst who speaks of the fraction of people who would not be excluded should report a nonexclusion rate that accounts for these inconclusive cases. Of course, the difference may be fairly small. In Pappas, a defense expert reported that the actual nonexclusion rate was still “99.3 percent of the Caucasian population.” Id. at 1105 (footnote omitted). See Kaye et al., supra note 83.
- Both samples heteroplasmic. In this third scenario, multiple sequences are seen in each sample. To keep track of things, we can call the sequences in the crime scene sample C1 and C2, and those in the suspect’s sample S1 and S2. If either C1 or C2 is very different from both S1 and S2, the suspect is excluded. If C1 and C2 are the same as S1 and S2, the suspect is included. Because detectable heteroplasmy is not very common, this inclusion is stronger evidence of identity than the simple match in the first scenario. Finally, in the middle range, where C1 is very close to S1 or S2, or C2 is very close to S1 or S2, the result is inconclusive.
A number of courts have rejected objections that the methods for mtDNA sequencing do not comport with Frye134 or Daubert135 and that the phenomenon of heteroplasmy or the limitations in the statistical analysis preclude the forensic use of this technology under either Rule 702 or Rule 403.136
Y chromosomes contain genes that result in development as a male rather than a female. Therefore, men are type XY and women are XX. A male child receives an X chromosome from his mother and a Y from his father; females receive two different X chromosomes, one from each parent. Like all chromosomes, the Y chromosome contains STRs and SNPs.
Because there is limited recombination between Y and X chromosomes, Y-STRs and Y-SNPs are inherited as a single block—a haplotype—from father to son. This means that the issues of population genetics and statistics are similar to those for mtDNA. No matter how many Y-STRs are in the haplotype, all the men in the same paternal line (up to the last mutation giving rise to a new line in the family tree) would match the crime scene sample.
134. E.g., Magaletti v. State, 847 So. 2d 523, 528 (Fla. Dist. Ct. App. 2003) (“[T]he mtDNA analysis conducted [on hair] determined an exclusionary rate of 99.93 percent. In other words, the results indicate that 99.93 percent of people randomly selected would not match the unknown hair sample found in the victim’s bindings.”); People v. Sutherland. 860 N.E.2d 178, 271–72 (Ill. 2006); People v. Holtzer, 660 N.W.2d 405, 411 (Mich. Ct. App. 2003); Wagner v. State, 864 A.2d 1037, 1043–49 (Md. Ct. Spec. App. 2005) (mtDNA sequencing admissible despite contamination and heteroplasmy).
135. E.g., United States v. Beverly, 369 F.3d 516, 531 (6th Cir. 2004) (“The scientific basis for the use of such DNA is well established.”); United States v. Coleman, 202 F. Supp. 2d 962, 967 (E.D. Mo. 2002) (“‘[a]t the most,’ seven out of 10,000 people would be expected to have that exact sequence of As, Ts, Cs, and Gs.”), aff’d, 349 F.3d 1077 (8th Cir. 2003); Pappas, 776 A.2d at 1095; State v. Underwood, 518 S.E.2d 231, 240 (N.C. Ct. App. 1999); State v. Council, 515 S.E.2d 508, 518 (S.C. 1999).
136. E.g., Beverly, 369 F.3d at 531 (“[T]he mathematical basis for the evidentiary power of the mtDNA evidence was carefully explained, and was not more prejudicial than probative.”); Pappas, 776 A.2d 1091.
Consequently, multiplication of allele frequencies is inappropriate, and an estimate of how many men might share the haplotype must be based on the frequency of that one haplotype in a relevant population. Population structure is a concern, and obtaining a suitable sample to estimate the frequency in a local population could be a challenge. If such a database is not available, DNA analysts might consider limiting their testimony on direct examination to the size of the available database, the population sampled, and the number of individuals in the database who share the crime scene haplotype. This presentation is less ambitious than a random-match probability, and courts must decide whether it gives the jury sufficient information to fairly assess the probative value of the match, which could be substantial.
When a standard DNA profile (involving a reasonable number of STRs or other polymorphisms of the other chromosomes) is available, there is little reason to add a Y-STR test. The profiles already are extremely rare. In some cases, however, standard STR typing will fail. Consider, for example, what happens when a PCR primer that targets an STR locus on, say, chromosome 16 is applied to a sample that contains a small number of sperm (from, say, a vasectomized man) and a huge number of cells from a woman who is a victim of sexual assault. Almost never will the primer lock onto the man’s chromosome 16. Therefore, his alleles on this chromosome will not produce a detectable peak in an electropherogram. But a primer for a Y-STR will not bind to the victim’s chromosomes—her chromosomes swamp the sample, but they are essentially invisible to the Y-STR primer. Because this primer binds only to the Y chromosomes from the man, only his STRs will be amplified. This is one example of how Y-STR profiling can be valuable in dealing with a mixture of DNA from several individuals. The next section provides other examples and describes other ways in which analysis of the Y chromosome can be valuable in mixture cases.
Although the statistics and population genetics of Y-STRs are different from the other STRs, the underlying technology for obtaining the profile is the same. On this basis, some courts have upheld the admission of these markers.137
Samples of biological trace evidence recovered from crime scenes often contain a mixture of fluids or tissues from different individuals. Examples include vaginal swabs collected as sexual assault evidence and bloodstain evidence from scenes where several individuals shed blood. However, not all mixed samples produce mixed STR profiles.138 Consider a sample in which 99% of the DNA comes from
137. E.g., Shabazz v. State, 592 S.E.2d 876, 879 (Ga. Ct. App. 2004); Curtis v. State, 205 S.W.3d 656, 660–61 (Tex. Ct. App. 2006).
138. The discussion in this section is limited to electropherograms of STR alleles. A recent paper reports a statistical technique that compares the known SNP genotypes (involving hundreds of
the defendant and 1% comes from a different individual. Even if some of the molecules from the minor contributor come in contact with the polymerase and an STR is amplified, the resulting signal might be too small to be detected—the peak in an electropherogram will blend into the background. Because the vast bulk of the amplified STRs will come from the defendant’s DNA, the electropherogram should show only one STR profile. In these situations, the interpretation of the single DNA profile is the same as when 100% of the DNA molecules in the sample are the defendant’s.
When the mixtures are more evenly balanced among contributors, however, the STRs from multiple contributors can appear as “extra” peaks. As a rule, because DNA from a single individual can have no more than two alleles at each locus,139 the presence of three or more peaks at several loci indicates that a mixture of DNA is in the sample.140Figure 6 shows another electropherogram from DNA recovered in People v. Pizarro.141 The fact that there are as many as four alleles at some loci and that many of the peaks match the victim’s) suggests that the sample is a mixture of the victim’s and another person’s DNA. Furthermore, a peak at the amelogenein locus shows that male DNA is part of the mixture. Because all the peaks that do not match the victim are part of the defendant’s STR profile, the mixture is consistent with the state’s theory that the defendant raped the victim.
Five approaches are available to cope with detectable mixtures. First, if a laboratory has other samples that do not show evidence of mixing, it can avoid the problem of deciphering the convoluted set of profiles. Even across a single stain, the proportions of a mixture can vary, and it might be possible to extract a DNA sample that does not produce a mixed signal.
Second, a chemical procedure exists to separate the DNA from sperm from a rape victim’s vaginal epithelial cell DNA.142 When this procedure works, the
thousands of SNPs) of a set of individuals to the SNPs detected in complex mixtures. The report states that the technique is able to discern “whether an individual is within a series of complex mixtures (2 to 200 individuals) when the individual contributes trace levels (at and below 1%) of the total genomic DNA.” Nils Homer et al., Resolving Individuals Contributing Trace Amounts of DNA to Highly Complex Mixtures Using High-Density SNP Genotyping Microarrays, 4 PLoS Genetics No. 8 (2008), available at http://www.plosgenetics.org/article/info%3Adoi%2F10.1371%2Fjournal.pgen.1000167.
139. This follows from the fact that individuals inherit chromosomes in pairs, one from each parent. An individual who inherits the same allele from each parent (a homozygote) can contribute only that one allele to a sample, and an individual who inherits a different allele from each parent (a heterozygote) will contribute those two alleles. Finding three or more alleles at several loci therefore indicates a mixture.
140. On rare occasions, an individual exhibits a phenotype with three alleles at a locus. This can be the result of a chromosome anomaly (such as a duplicated gene on one chromosome or a mutation). A sample from such an individual is usually easily distinguished from a mixed sample. The three-allele variant is seen at only the affected locus, whereas with mixtures, more than two alleles typically are evident at several loci.
141. See supra Figure 5.
142. The nucleus of a sperm cell lies behind a protective structure that does not break down as easily as the membrane in an epithelial cell. This makes it possible to disrupt the epithelial cells first and extract their DNA, and then to use a harsher treatment to disrupt the sperm cells.
Figure 6. Electropherogram in People v. Pizarro that can be interpreted as a mixture of DNA from the victim and the defendant.

Source: Steven Myers and Jeanette Wallin, California Department of Justice, provided the image.
laboratory can assign the DNA profiles to the different individuals because it has created, in effect, two samples that are not mixtures.
Third, in sexual assault cases, Y chromosome testing can reveal the number of men (from different paternal lines) whose DNA is being detected and whether the defendant’s Y chromosome is consistent with his being in one of these paternal lines.143 Because only males have Y chromosomes, the female DNA in a mixture has no effect.
Fourth, a laboratory simply can report that a defendant’s profile is consistent with the mixed profile, and it can provide an estimate of the proportion of the relevant population that also cannot be excluded (or would be included).144 When
143. E.g., State v. Polizzi, 924 So. 2d 303, 308–09 (La. Ct. App. 2006) (testing for Y-STRs on “the genital swab with the DNA profile from the Defendant’s buccal swab,…the Defendant or any of his paternal relatives could not be excluded as having been a donor to the sample from the victim,” while “99.7 percent of the Caucasian population, 99.8 percent of the African American population, and 99.3 percent of the Hispanic population could be excluded as donors of the DNA in the sample.”).
144. E.g., State v. Roman Nose, 667 N.W.2d 386, 394 n.5 (Minn. 2003). If the laboratory can explain why one or more of the defendant’s alleles do not appear in the mixed profile from the
an individual’s DNA—for example, the victim’s—is known to be in a two-person crime scene sample, the profile of the unknown person is readily deduced. In those situations, the analysis of a remaining single-person profile can proceed in the ordinary fashion.
Finally, a laboratory can try to determine (or make assumptions about) how many contributors are present and then deduce which set of alleles is likely to be from each contributor. To accomplish this, DNA analysts look to such clues as the number of peaks in an expected allele-size range and the imbalance in the heights of the peaks.145 A good deal of judgment can go into the determination of which peaks are real, which are artifacts, which are “masked,” and which are absent for some other reason.146 Courts generally have rejected arguments that mixture analysis is so unreliable or so open to manipulation that the results are inadmissible.147 In addition, expert computer systems have been devised for facilitating the analysis and for automatically “deconvoluting” mixtures.148 Once they are validated, these systems can make the process more standardized.
The five approaches listed here are not mutually exclusive (and not all apply to every case). When the number of contributors to a mixture is in doubt, for example, a laboratory is not limited to giving the overall probability of excluding (or including) an individual as a possible contributor (the statistic mentioned as part of the fourth method). The 1996 NRC report observed that “when the contributors to a mixture are not known or cannot otherwise be distinguished, a likelihood-ratio approach offers a clear advantage [over the simplistic exclusion-inclusion statistic] and is particularly suitable.”149 Despite the arguments of some
crime scene, it might be willing to declare a match not withstanding this discrepancy. Of course, as the number of alleles that must be present for there to be a match declines, the proportion of the population that would be included goes up.
145. See, e.g., Roberts v. United States, 916 A.2d 922, 932–35 (D.C. 2007) (holding such inferences to be admissible).
146. The proportion of the population included in a mixture and the likelihood ratios conditioned on a particular genotype do not take into account the other possible genotypes that the expert eliminated in a subjective analysis. William C. Thompson, Painting the Target Around the Matching Profile: The Texas Sharpshooter Fallacy in Forensic DNA Interpretation, 8 Law, Probability & Risk 257 (2009). Adhering to preestablished standards and protocols for interpreting mixtures reduces the range of judgment in settling on the most likely set of genotypes to consider. Recent recommendations appear in Bruce Budowle et al., Mixture Interpretation: Defining the Relevant Features for Guidelines for the Assessment of Mixed DNA Profiles in Forensic Casework, 54 J. Forensic Sci. 810 (2009) (with commentary at 55 J. Forensic Sci. 265 (2010)); Peter Gill et al., National Recommendations of the Technical UK DNA Working Group on Mixture Interpretation for the NDNAD and for Court Going Purposes, 2 Forensic Sci. Int’l Genetics 76 (2008).
147. Roberts, 916 A.2d at 932 n.9 (citing cases).
148. See, e.g., Tim Clayton & John Buckleton, Mixtures, in Forensic DNA Evidence Interpretation 217 (John Buckleton et al. eds., 2005); Mark W. Perlin et al., Validating TrueAllele® DNA Mixture Interpretation, 56 J. Forensic Sci. (forthcoming 2011).
149. NRC II, supra note 9, at 129.
legal commentators that likelihood ratios are inherently prejudicial,150 and despite objections based on Frye or Daubert, almost all courts have found likelihood ratios admissible in mixture cases.151
D. Offender and Suspect Database Searches
States and the federal government are amassing huge databases consisting of the DNA profiles of suspected or convicted offenders.152 If the DNA profile from a crime scene stain matches one of those on file, the person identified by this “cold hit” will become the target of the investigation. Prosecution may follow.
These database-trawl cases can be contrasted with traditional “confirmation cases” in which the defendant already was a suspect and the DNA testing provided additional evidence against him. In confirmation cases, statistics such as the estimated frequency of the matching DNA profile in various populations, the equivalent random-match probabilities, or the corresponding likelihood ratios can be used to indicate the probative value of the DNA match.153
In trawl cases, however, an additional question arises—does the fact that the defendant was selected for prosecution by trawling require some adjustment to the usual statistics? The legal issues are twofold. First, is a particular quantity—be it the unadjusted random-match probability or some adjusted probability—scientifically valid (or generally accepted) in the case of a database search? If not, it must be excluded under the Daubert (or Frye) standards. Second, is the statistic irrelevant or unduly misleading? If so, it must be excluded under the rules that
150. E.g., William C. Thompson, DNA Evidence in the O.J. Simpson Trial, 67 U. Colo. L. Rev. 827, 855–56 (1996); see also R.C. Lewontin, Population Genetic Issues in the Forensic Use of DNA, in 1 Modern Scientific Evidence: The Law and Science of Expert Testimony § 17-5.0, at 703–05 (Faigman et al. eds, 1st ed. 1998).
151. E.g., State v. Garcia, 3 P.3d 999 (Ariz. Ct. App. 1999) (likelihood ratios admissible under Frye to explain mixed sample); Commonwealth v. Gaynor, 820 N.E.2d 233, 252 (Mass. 2005) (“Likelihood ratio analysis is appropriate for test results of mixed samples when the primary and secondary contributors cannot be distinguished…. It need not be applied when a primary contributor can be identified.”) (citation omitted); People v. Coy, 669 N.W.2d 831, 835–39 (Mich. Ct. App. 2003) (incorrectly treating mixed-sample likelihood ratios as a part of the statistics on single-source DNA matches that had already been held to be generally accepted); State v. Ayers, 68 P.3d 768, 775 (Mont. 2003) (affirming trial court’s admission of expert testimony where expert used likelihood ratios to explain DNA results from a sample known to contain a mixture of DNA); cf. Coy v. Renico, 414 F. Supp. 2d 744, 762–63 (E.D. Mich. 2006) (stating that the use of likelihood ratio and other statistics for a mixed stain in People v. Coy, supra, was sufficiently accepted in the scientific community to be consistent with due process).
152. See supra Section II.E.
153. On the computation and admissibility of such statistics, see supra Section IV.
require all evidence to be relevant and not unfairly prejudicial. To clarify, we summarize the statistical literature on this point. Then, we describe the emerging case law.
a. The statistical analyses of adjustment
All statisticians agree that, in principle, the search strategy affects the probative value of a DNA match. One group describes and emphasizes the impact of the database match on the hypothesis that the database does not contain the source of the crime scene DNA. This is a “frequentist” view. It asks how frequently searches of innocent databases—those for which the true source is someone outside the database—will generate cold hits. From this perspective, trawling is a form of “data mining” that produces a “selection effect” or “ascertainment bias.” If we pick a lottery ticket at random, the probability p that we have the winning ticket is negligible. But if we search through all the tickets, sooner or later we will find the winning one. And even if we search through some smaller number N of tickets, the probability of picking a winning ticket is no longer p, but Np.154 Likewise, if DNA from N innocent people is examined to determine if any of them match the crime scene DNA, then the probability of a match in this group is not p, but some quantity that could be as large as Np. This type of reasoning led the 1996 NRC committee to recommend that “[w]hen the suspect is found by a search of DNA databases, the random-match probability should be multiplied by N, the number of persons in the database.”155 The 1992 committee156 and the FBI’s former DNA Advisory Board157 took a similar position.
154. The analysis of the DNA database search is more complicated than the lottery example suggests. In the simple lottery, there was exactly one winner. The trawl case is closer to a lottery in which we hold a ticket with a winning number, but it might be counterfeit, and we are not sure how many counterfeit copies of the winning ticket were in circulation when we bought our N tickets.
155. NRC II, supra note 9, at 161 (Recommendation 5.1).
156. Initially, the board explained that
Two questions arise when a match is derived from a database search: (1) What is the rarity of the DNA profile? and (2) What is the probability of finding such a DNA profile in the database searched? These two questions address different issues. That the different questions produce different answers should be obvious. The former question addresses the random match probability, which is often of particular interest to the fact finder. Here we address the latter question, which is especially important when a profile found in a database search matches the DNA profile of an evidence sample.
DNA Advisory Board, Statistical and Population Genetics Issues Affecting the Evaluation of the Frequency of Occurrence of DNA Profiles Calculated from Pertinent Population Database(s), 2 Forensic Sci. Comm., July 2000, available at http://www.fbi.gov/hq/lab/fsc/backissu/july2000/dnastat.htm. After a discussion of the literature as of 2000, the Board wrote that “we continue to endorse the recommendation of the NRC II Report for the evaluation of DNA evidence from a database search.”
157. The first NRC committee wrote that “[t]he distinction between finding a match between an evidence sample and a suspect sample and finding a match between an evidence sample and one of many entries in a DNA profile databank is important.” It used the same Np formula in a numerical example to show that “[t]he chance of finding a match in the second case is considerably higher,
No one questions the mathematics that show that when the database size N is very small compared with the size of the population, Np is an upper bound on the expected frequency with which searches of databases will incriminate innocent individuals when the true source of the crime scene DNA is not represented in the databases. The “Bayesian” school of thought, however, suggests that the frequency with which innocent databases will be falsely accused of harboring the source of the crime scene DNA is basically irrelevant. The question of interest to the legal system is whether the one individual whose database DNA matches the trace-evidence DNA is the source of that trace. As the size of a database approaches that of the entire population, finding one and only one matching individual should be more, not less, convincing evidence against that person. Thus, instead of looking at how surprising it would be to find a match in a large group of innocent suspects, this school of thought asks how much the result of the database search enhances the probability that the individual so identified is the source. The database search is actually more probative than the confirmation search because the DNA evidence in the trawl case is much more extensive. Trawling through large databases excludes millions of people, thereby reducing the number of people who might have left the trace evidence if the suspect did not. This additional information increases the likelihood that the defendant is the source, although the effect is indirect and generally small.158
Of course, when the cold hit is the only evidence against the defendant, the total package of evidence in the trawl case is less than in the confirmation case. Nonetheless, the Bayesian treatment shows that the DNA part of the total evidence is more powerful in a cold-hit case because this part of the evidence is more complete than when the search for matching DNA is limited to a single suspect. This reasoning suggests that the random-match probability (or, equivalently, the frequency p in the population) understates the probative value of the unique DNA match in the trawl case. And if this is so, then the unadjusted random-match probability or frequency p can be used as a conservative indication of the probative value of the finding that, of the many people in the database, only the defendant matches.159
because one…fishes through the databank, trying out many hypotheses.” NRC I, supra note 8, at 124. Rather than proposing a statistical adjustment to the match probability, however, that committee recommended using only a few loci in the databank search, then confirming the match with additional loci, and presenting only “the statistical frequency associated with the additional loci….” Id. at 124 tbl. 1.1.
158. When the size of the database approaches the size of the entire population, the effect is large. Finding that only one individual in a large database has a particular profile also raises the probability that this profile is very rare, further enhancing the probative value of the DNA evidence.
159. This analysis was developed by David Balding and Peter Donnelly. For informal expositions, see, for example, Peter Donnelly & Richard D. Friedman, DNA Database Searches and the Legal Consumption of Scientific Evidence, 97 Mich. L. Rev. 931 (1999); Kaye, supra note 109; Simon Walsh & John Buckleton, DNA Intelligence Databases, in Forensic DNA Evidence Interpretation 439 (John Buckleton et al. eds., 2005). For a related analysis directed at the average probability that an individual
b. The judicial opinions on adjustment
The need for an adjustment has been vigorously debated in the statistical, and to a lesser extent, the legal literature.160 The dominant view in the journal articles is that the random-match probability or frequency need not be inflated to protect the defendant. The major opinions to confront this issue agree that p is admissible against the defendant in a trawl case.161 They reason that all the statistics are admissible under Frye and Daubert because there is no controversy over how they are computed. They then assume that both p and Np are logically relevant and not prejudicial in a trawl case.
But commentators have pointed out that if the frequentist position that trawling degrades the probative value of the match is correct, then it is hard to see what p offers the jury. Conversely, if the Bayesian position that trawling enhances the probative value of the match is correct, then it is hard to see what Np offers the jury.162 Thus, it has been argued that, to decide whether p should be admissible when offered by the prosecution and whether Np (or some variant of it) should be admissible when offered by the defense, the law needs to directly confront the schism between the frequentist and Bayesian perspectives on characterizing the probative value of a cold hit.163
2. Near-miss (familial) searching
Normally, police trawl a DNA database to see if any recorded STR profiles match a crime scene profile. It is not generally necessary to inform the jury that the defendant was located in this manner. Indeed, the rules of evidence sometimes prohibit this proof over the objection of the defendant.164 Another search pro-
identified through a database trawl is the source of a crime scene DNA sample, see Yun S. Song et al., Average Probability That a “Cold Hit” in a DNA Database Search Results in an Erroneous Attribution, 54 J. Forensic Sci. 22, 23–24 (2009).
160. For citations to this literature, see Kaye, supra note 109; Walsh & Buckleton, supra note 159, at 464.
161. People v. Nelson, 185 P.3d 49 (Cal. 2008); United States v. Jenkins, 887 A.2d 1013 (D.C. 2005). The cases are analyzed in Kaye, supra note 109.
162. Furthermore, even if an adjustment is logically required, Np might be too extreme because the offender databases include the profiles of individuals—those in prison at the time of the offense, for instance—who could not have been the source of the crime scene sample. To that extent, Np overstates the expected frequency of matches to innocent individuals in the database. Kaye, supra note 109; David H. Kaye, People v. Nelson: A Tale of Two Statistics, 7 Law, Probability & Risk 249 (2008).
163. Kaye, supra note 109; Kaye, supra note 162.
164. The common law of evidence and Federal Rule of Evidence 404 prevent the government from proving that a defendant has committed other crimes when the only purpose of the revelation is to suggest a general propensity toward criminality. See, e.g., 1 McCormick on Evidence, supra note 3, § 190. Proof that the defendant was identified through a database search is likely to suggest the existence of a criminal record, because it is widely known that existing law enforcement DNA databases are largely filled with the profiles of convicted offenders. Nonetheless, where the bona fide and important purpose of the disclosure is “to complete the story” (id.) or to help the jury to understand an Np
cedure can lead to charges against a defendant who is not even in the database. The clearest illustration is the case of identical twins, one of whom is a convicted offender whose DNA type is on file and the other who has never been typed. If the convicted twin was in prison when the crime under investigation was committed, and if the police realized that he had an identical twin, suspicion should fall on the identical twin. Presumably, the police would seek a sample from this twin, and at trial it would not be necessary for the prosecution to explain the roundabout process through which he was identified.
In this example, the defendant was found because a relative’s DNA led the police to him. More generally, the fact that close relatives share more alleles than other members of the same subpopulation can be exploited as an investigative tool. Rather than search for a match at all 13 loci in an STR profile, police could search for a near miss—a partial match that is much more probable when the partly matching profile in the database comes from a close relative than when it comes from an unrelated person. (Analysis of Y-STRs or mtDNA then could determine whether the offender who provided the partially matching DNA in the database probably is in the same paternal or maternal lineage as the unknown individual who left DNA at the scene of the crime.)
Such “familial searching” raises technical and policy questions. The technical and practical challenge is to devise a search strategy that keeps the number of false leads to a tolerable level. The policy question is whether exposing relatives to the possibility of being investigated on the basis of genetic leads from their kin is appropriate.165
In receiving the DNA evidence, courts might consider having the prosecution describe the match without revealing that the defendant’s close relative is a known or suspected criminal. In addition, if database trawls degrade the probative value of a perfect match in the database—a theory discussed in the previous subsection—then the usual random-match probability or estimated frequency exaggerates the value of the match derived from a database search. From the frequentist perspective, one must ask how often trawling databases for leads to individuals (both within and outside the database) will produce false accusations. From the Bayesian perspective, however, the usual match probabilities and likeli-
statistic, Rule 404 itself arguably does not prevent the prosecution (and certainly not the defense) from revealing that the defendant was found through a DNA database trawl. In the absence of a categorical rule of exclusion (like the one in Rule 404), a case-by-case balancing of the value of the information for its legitimate purposes as against its potential prejudice to the defendant is required. See id.
165. See, e.g., Bruce Budowle et al., Clarification of Statistical Issues Related to the Operation of CODIS, in Genetic Identity Conference Proceedings: 18th Int’l Symposium on Human Identification (2006), available at http://www.promega.com/GENETICIDPROC/ussymp17proc/oralpresentations/budowle.pdf; Henry T. Greely et al., Family Ties: The Use of DNA Offender Databases to Catch Offenders’ Kin, 34 J.L. Med. & Ethics 248 (2006); Erica Haimes, Social and Ethical Issues in the Use of Familiar Searching in Forensic Investigations: Insights from Family and Kinship Studies, 34 J.L. Med. & Ethics 262 (2006).
hoods can be used because, if anything, they understate the probative value of the DNA information.166
3. All-pairs matching within a database to verify estimated random-match probabilities
A third and final use of police intelligence databases has evidentiary implications. Section IV.E explained how population genetics models and reference samples for determining allele frequencies are used to estimate DNA genotype frequencies. Large databases can be used to check these theoretical computations. In New Zealand, for example, researchers compared every six-locus STR profile in the national database with every other profile.167 At the time, there were 10,907 profiles. This means that there were about 59 million distinct pairs.168 Because the theoretical random-match probability was about 1 in 50 million, if all the individuals represented in the database were unrelated, one would expect that an exhaustive comparison of the profiles for these 59 million pairs would produce only about one match. In fact, the 59 million comparisons revealed 10 matches. The excess number of matches is evidence that not all the individuals in the database were unrelated, that the true match probability was smaller than the theoretical calculation, or both. In fact, eight of the pairs were twins or brothers. The ninth was a duplicate (because one person gave a sample as himself and then again pretending to be someone else). The tenth was apparently a match between two unrelated people. This exercise thus confirmed the theoretical computation of the random-match probability. On average, the theoretical match probability was about 1/50,000,000, and the rate of matches in the unrelated pairs within the database was 1/59,000,000.
In the United States, defendants have sought discovery of the criminal-offender databases to determine whether the number of matching and partially matching pairs exceeds the predictions made with the population genetics model.169 An early report about partial matches in a state database in Arizona was said to show extraordinarily large numbers of partial matches (without accounting for the combinatorial explosion in the number of comparisons in an all-pairs data-
166. See Kaye, supra note 3; supra Section V.D.1.
167. Walsh & Buckleton, supra note 159, at 463.
168. Altogether, nearly 11,000 people were represented in the New Zealand database. Hence, about 10,907 × 10,907 pairs such as (1,1), (1,2), (1,3),…, (1,10907), (2,1), (2,2), (2,3),…, (2,10907),…(10907,1), (10907,2), (10907,3), (10907,10907) can be formed. This amounts to almost 119 million possible pairs. Of course, there is no point in checking the pairs (1,1), (2,2),…(10907,10907). Thus, the number of ordered pairs with different individuals is 119 million minus a mere 10,907. The subtraction hardly changes anything. Finally, ordered pairs such as (1,5) and (5,1) involve the same two people. Therefore, the number of distinct pairs of people is about half of 119 million—the 59 million figure in the text.
169. Jason Felch & Maura Dolan, How Reliable Is DNA in Identifying Suspects? L.A. Times, July 20, 2008.
base search).170 However, some scientists question the utility of the investigative databases for population genetics research.171 They observe that these databases contain an unknown number of relatives, that they might contain duplicates, and that the population in the offender databases is highly structured. These complicating factors would need to be considered in testing for an excess of matches or partial matches. Studies of offender databases in Australia and New Zealand that make adjustments for population structure and close relatives have shown substantial agreement between the expected and observed numbers of partial matches, at least up to the nine STR loci used in those databases.172
The existence of large databases also provides a means of estimating a random-match probability without making any modeling assumptions. For the New Zealand study, even ignoring the possibility of relatives and duplicates, there were only 10 matches out of 59 million comparisons. The empirical estimate of the random-match probability is therefore about 1 in 5.9 million. This is about 10 times larger than the theoretical estimate, but still quite small. As this example indicates, crude but simple empirical estimates from all-pairs comparisons in large databases may well produce random-match probabilities that are larger than the theoretical estimates (as expected when full siblings or other close relatives are in the databases), but the estimated probabilities are likely to remain impressively small.
170. Id.; Kaye, supra note 3. As illustrated supra note 168, an all-pairs search in a large database of size N will involve N(N − 1)/2, or about N2/2 comparisons. For example, a database of 6 million samples gives rise to some 18,000,000,000,000 comparisons. Even with no population structure, relatives, and duplicates, and with random-match probabilities in the trillionths, one would expect to find a large number of matches or near-matches. An analogy can be made to the famous “birthday problem” mentioned in the 1996 NRC Report, supra note 9, at 165. In its simplest form, the birthday problem assumes that equal numbers of people are born every day of the year. The problem is to determine the minimum number of people in a room such that the odds favor there being at least two of them who were born on the same day of the same month. Focusing solely on the random-match probability of 1/365 for a specified birthday makes it appear that a huge number of people must be in the room for a match to be likely. After all, the chance of a match between two individuals having a given birthday (say, January 1) is (ignoring leap years) a miniscule 1/365 × 1/365 = 1/133,225. But because the matching birthday can be any one of the 365 days in the year and because there are N(N − 1)/2 ways to have a match, it takes only N = 23 people before it is more likely than not that at least two people share a birthday. The birthday problem thus shows that surprising coincidences commonly occur even in relatively small databases. See, e.g., Persi Diaconis & Frederick Mosteller, Methods for Studying Coincidences, 84 J. Am. Statistical Ass’n 853 (1989).
171. Bruce Budowle et al., Partial Matches in Heterogeneous Offender Databases Do Not Call into Question the Validity of Random Match Probability Calculations, 123 Int’l J. Legal Med. 59 (2009).
172. James M. Curran et al., Empirical Support for the Reliability of DNA Evidence Interpretation in Australia and New Zealand, 40 Australian J. Forensic Sci. 99, 102–06 (2008); Bruce S. Weir, The Rarity of DNA Profiles, 1 Annals Applied Stat. 358 (2007); B.S. Weir, Matching and Partially-Matching DNA Profiles, 49 J. Forensic Sci. 1009, 1013 (2004); cf. Laurence D. Mueller, Can Simple Population Genetic Models Reconcile Partial Match Frequencies Observed in Large Forensic Databases? 87 J. Genetics (India) 101 (2008) (maintaining that excess partial matches in an Arizona offender database are not easily reconciled with theoretical expectations). This literature is reviewed in David H. Kaye, Trawling DNA Databases for Partial Matches: What Is the FBI Afraid of? 19 Cornell J.L. & Pub. Pol’y 145 (2009).
Most routine applications of DNA technology in the forensic setting involve the identification of human beings—suspects in criminal cases, missing persons, or victims of mass disasters. However, inasmuch as DNA analysis might be informative in any kind of case involving biological material, DNA analysis has found application in such diverse situations as identification of individual plants and animals that link suspects to crime scenes, enforcement of endangered species and other wildlife regulations, investigation of patent issues involving specific animal breeds and plant cultivars, identification of fraudulently labeled foodstuffs, identification of sources of bacterial and viral epidemic outbreaks, and identification of agents of bioterrorism.173 These applications are directed either at identifying the species origin of an item or at distinguishing among individuals (or subgroups) within a species. In deciding whether the evidence is scientifically sound, it can be important to consider the novelty of the application, the validity of the underlying scientific theory, the validity of any statistical interpretations, and the relevant scientific community to consult in assessing the application. This section considers these factors in the context of nonhuman DNA testing.
Evolution is a branching process. Over time, populations may split into distinct species. Ancestral species and some or all of their branches become extinct. Phylogenetics uses DNA sequences to elucidate these evolutionary “trees.” This information can help determine the species of the organism from which material has been obtained. For example, the most desirable Russian black caviar originates from three species of wild sturgeon inhabiting the Volga River–Caspian Sea basin. But caviar from other sturgeon species is sometimes falsely labeled as originating from these three species—in violation of food labeling laws. Moreover, the three sturgeon species are listed as endangered, and trade in their caviar is restricted. A test of caviar species based on DNA sequence variation in a mitochondrial gene found that 23% of caviar products in the New York City area were mislabeled,174 and in United States v. Yazback, caviar species testing was used to convict an
173. See, e.g., R.G. Breeze et al., Microbial Forensics (2005); Laurel A. Neme, Animal Investigators: How the World’s First Wildlife Forensics Lab Is Solving Crimes and Saving Endangered Species (2009). In still other situations, DNA testing has been used to establish the identity of a missing or stolen animal. E.g., Augillard v. Madura, 257 S.W.3d 494 (Tex. App. 2008) (action for conversion to recover dog lost during Hurricane Katrina); Guillermo Giovambattista et al., DNA Typing in a Cattle Stealing Case, 46 J. Forensic Sci. 1484 (2001).
174. Rob DeSalle & Vadim J. Birstein, PCR Identification of Black Caviar, 381 Nature 197 (1996); Vadim J. Birstein et al., Population Aggregation Analysis of Three Caviar-Producing Species of Sturgeons and Implications for the Species Identification of Black Caviar, 12 Conservation Biology 766 (1998).
importer of gourmet foods for falsely labeling fish eggs from the environmentally protected American paddlefish as the more prized Russian sevruga caviar.175
Phylogenetic analysis also is used to study changes in populations of organisms of the same species. In State v. Schmidt,176 a physician was convicted of attempting to murder his former lover by injecting her with the HIV virus obtained from an infected patient. The virus evolves rapidly—its sequence can change by as much as 1% per year over the course of infection in a single individual. In time, an infected individual will harbor new strains, but these will be more closely related to the particular strain (or strains) that originally infected the individual than to the diverse strains of the virus in the geographic area. The victim in Schmidt had fewer strains of HIV than the patient—indicating a later infection—and all the victim’s strains were closely related to a subset of the patient’s strains—indicating that the victim’s strains originated from that subset then in the patient. This technique of examining the genetic similarities and differences in two populations of viruses has been used in other cases across the world.177
The FBI employed similar reasoning to conclude that the anthrax spores in letters sent through the mail in 2001 came from the descendants of bacteria first cultured from an infected cow in Texas in 1981. This “Ames strain” was disseminated to various research laboratories over the years, and the FBI also attempted to associate the letter spores with particular collections of anthrax bacteria (all derived from the one Ames strain) now housed in different laboratories.178
Both the caviar and the HIV cases exemplify the translation of established scientific methods into a forensic application. The mitochondrial gene used for species identification in Yazback was the cytochrome b gene. Having accumulated mutations over time, this gene commonly is used for assessing species relationships among vertebrates, and the database of cytochrome b sequences is extensive. In particular, this gene sequence previously had been used to determine the evolutionary placement of sturgeons among other species of fish.179 Likewise, the use of phylogenetic analysis for assessing relationships among HIV strains has provided critical insights into the biology of this deadly virus.
175. Dep’t of Justice, Caviar Company and President Convicted in Smuggling Conspiracy, available at http://www.usdoj.gov/opa/pr/2002/January/02_enrd_052.htm. An earlier case is described in Andrew Cohen, Sturgeon Poaching and Black Market Caviar: A Case Study, 48 J. Env’l Biology Fishes 423 (1997).
176. 699 So. 2d 448 (La. Ct. App. 1997) (holding that the evidence satisfied Daubert).
177. Edwin J Bernard et al., The Use of Phylogenetic Analysis as Evidence in Criminal Investigation of HIV Transmission, Feb. 2007, available at http://www.nat.org.uk/Media%20library/Files/PDF%20Documents/HIV-Forensics.pdf.
178. See National Research Council, Committee on the Review of the Scientific Approaches Used During the FBI’s Investigation of the 2001 Bacillus Anthracis Mailings, Review of the Scientific Approaches Used During the FBI’s Investigation of the 2001 Anthrax Letters (2011).
179. Sturgeon Biodiversity and Conservation (Vadim J. Birstein et al. eds., 1997).
That said, how the phylogenetic analysis is implemented and the genes used for the analysis may prompt questions in some cases.180 Both the computer algorithm used to align DNA sequences prior to the construction of the phylogenetic tree and the computer algorithms used to build the tree contain assumptions that can influence the outcomes. Consequently, alignments generated by different software can result in different trees, and different tree-building algorithms can yield different trees from the same alignments. Thus, phylogenetic analysis should not be looked upon as a simple mechanical process. In Schmidt, the investigators anticipated and addressed potential problem areas in their choice of sequence data to collect and by using different algorithms for phylogenetic analysis. The results from the multiple analyses were the same, supporting the overall conclusion.181
DNA analysis to determine that trace evidence originated from a particular individual within a species requires both a valid analytical procedure for forensic samples and at least a rough assessment of how rare the DNA types are in the population. In human DNA testing, suitable reference databases permit reasonable estimates of allele frequencies among groups of human beings (see supra Section IV), but adequate databases will not always be available for other organisms. Nonetheless, a match between the DNA at a crime scene and the organism that could be the source of that trace evidence still may be informative. In these cases, a court may consider admitting testimony about the matching features along with circumscribed, qualitative explanations of the significance of the similarities.182
Such cases began appearing in the 1990s. In State v. Bogan,183 for example, a woman’s body was found in the desert, near several Palo Verde trees. A detective noticed two Palo Verde seed pods in the bed of a truck that the suspect was driving before the murder. However, genetic variation in Palo Verde tree DNA had not been widely studied, and no one knew how much variation actually
180. One cannot assume that cytochrome b gene testing, for example, is automatically appropriate for all species identification. Mitochondria are maternally inherited, and one can ask whether cross-breeding between different species of sturgeon could make a sturgeon of one species appear to be another species because it carries mitochondrial DNA originating from the other species. Mitochondrial introgression has been detected in several vertebrate species. Coyote mtDNA in wolves and cattle mtDNA in bison are notable examples. Introgression in sturgeon has been reported—some individual sturgeon appearing to be of one of the prized Volga region caviar species were found to carry cytochrome b genes from a lesser regarded non-Volga species. These examples indicate the need for specialized knowledge of the basic biology and ecology of the species in question.
181. On the need for caution in the interpretation of HIV sequence similarities, see Bernard et al., supra note 177.
182. See generally Kaye et al., supra note 1 (discussing various ways to explain “matches” in forensic identification tests).
183. 905 P.2d 515 (Ariz. Ct. App. 1995) (holding that the admission of the DNA match was proper under Frye).
existed within the species. Accordingly, a method for genetic analysis had to be developed, assessed for what it revealed about genetic variability, and evaluated for reliability. A university biologist chose random amplified polymorphic DNA (RAPD) analysis, a PCR-based method then commonly used for the detection of variation within species for which no genomic sequence information exists. This approach employs a single short piece of DNA with a random, arbitrary sequence as the PCR primer; the amplification products are DNA fragments of unknown sequence and variable length that can be separated by electrophoresis into a barcode-like “fingerprint” pattern. In a blind trial, the biologist was able to show that the DNA from nearly 30 Palo Verde trees yielded distinct RAPD patterns.184 He testified that the two pods “were identical” and “matched completely with” a particular tree and “didn’t match any of the [other] trees.” In fact, he went so as to say that he felt “quite confident in concluding that” the tree’s DNA would be distinguishable from that of “any tree that might be furnished” to him. Numerical estimates of the random-match probability were not introduced.185
The first example of an animal identification using STR typing involved linking evidence cat hairs to a particular cat. In R. v. Beamish, a woman disappeared from her home on Prince Edward Island, on Canada’s eastern seaboard. Weeks later a man’s brown leather jacket stained with blood was discovered in a plastic bag in the woods. In the jacket’s lining were white cat hairs. After the missing woman’s body was found in a shallow grave, her estranged common-law husband was arrested and charged with murder. He lived with his parents and a white cat named Snowball. A laboratory already engaged in the study of genetic diversity in cats showed that the DNA profile of the evidence cat hairs matched Snowball at 10 STR loci. Based on a survey of genetic variation in domestic cats generated for this case, the probability of a chance match was offered as evidence in support of the hypothesis that the hairs originated from Snowball.186
184. He analyzed samples from the nine trees near the body and another 19 trees from across the county. He “was not informed, until after his tests were completed and his report written, which samples came from” which trees. Id. at 521. Furthermore, unbeknownst to the experimenter, two apparently distinct samples were prepared from the tree at the crime scene that appeared to have been abraded by the defendant’s truck. The biologist correctly identified the two samples from the one tree as matching, and he “distinguished the DNA from the seed pods in the truck bed from the DNA of all twenty-eight trees except” that one. Id.
185. RAPD analysis does not provide systematic information about sequence variation at defined loci. As a result, it is not possible to make a reliable estimate of allele or genotype frequencies at a locus, nor can one make the assumption of genetic independence required to legitimately multiply frequencies across multiple loci, as one can with STR markers. Furthermore, RAPD profile results are not generally portable between laboratories. Often, profiles generated by different laboratories will differ in their details. Therefore, RAPD profile data are not amenable to the generation of large databases. Nonetheless, the state’s expert estimated a random match probability of 1 in 1,000,000, and the defense expert countered with 1 in 136,000. The trial court excluded both estimates because of the then-existing controversy (see Section IV) over analogous estimates for human RFLP genotypes.
186. See Marilyn A. Menott-Haymond et al., Pet Cat Hair Implicates Murder Suspect, 386 Nature 774 (1997).
In Beamish, there was no preexisting population database characterizing STR polymorphism in domestic cats, but the premise that cats exhibit substantial genetic variation at STR loci was in accord with knowledge of STR variation in other mammals. Moreover, testing done on two small cat populations provided evidence that the STR loci chosen for analysis were polymorphic and behaved as independent genetic characteristics, allowing allele frequency estimates to be used for the calculation of random-match probabilities as is done with human STR data. On this basis, the random-match probability for Snowball’s STR profile was estimated to be one in many millions, and the trial court admitted this statistic.187
An animal-DNA random-match probability prompted a reversal, however, in a Washington case. In State v. Leuluaialii,188 the prosecution offered testimony of an STR match with a dog’s blood that linked the defendants to the victims’ bodies. The defendants objected, seeking a Frye hearing, but the trial court denied this motion and admitted testimony that included the report that “the probability of finding another dog with Chief’s DNA profile was 1 in 18 billion [or] 1 in 3 trillion.”189 The state court of appeals remanded the case for a hearing on general acceptance, cautioning that “[b]ecause PE Zoogen has not yet published sufficient data to show that its DNA markers and associated probability estimates are reliable, we would suggest that other courts tread lightly in these waters and closely examine canine DNA results before accepting them at trial.”190
The scientific literature shows continued use of STR profiling191 (as well as the use of SNP typing)192 to characterize individuals in plant and animal populations. STR databases have been established for domestic and agriculturally significant animals such as dogs, cats, cattle, and horses as well as for a number of plant species.193 Critical to the use of these databases is an understanding of the
187. David N. Leff, Killer Convicted by a Hair: Unprecedented Forensic Evidence from Cat’s DNA Convinced Canadian Jury, Bioworld Today, Apr. 24, 1997, available in 1997 WL 7473675 (“the frequency of the match came out to be on the order of about one in 45 million,” quoting Steven O’Brien); All Things Considered: Cat DNA (NPR broadcast, Apr. 23, 1997), available in 1997 WL 12832754 (“it was less than one in two hundred million,” quoting Steven O’Brien).
188. 77 P.3d 1192 (Wash. Ct. App. 2003).
189. Id. at 1196.
190. Id. at 1201.
191. E.g., Kathleen J. Craft et al., Application of Plant DNA Markers in Forensic Botany: Genetic Comparison of Quercus Evidence Leaves to Crime Scene Trees Using Microsatellites, 165 Forensic Sci. Int’l 64 (2007) (differentiation of oak tree leaves); Christine Kubik et al., Genetic Diversity in Seven Perennial Ryegrass (Lolium perenne L.) Cultivars Based on SSR Markers, 41 Crop Sci. 1565 (2001) (210 ryegrass samples correctly assigned to seven cultivars).
192. E.g., Bridgett M. vonHoldt et al., Genome-wide SNP and Haplotype Analyses Reveal a Rich History Underlying Dog Domestication, 464 Nature 898 (2010) (48,000 SNPs in 912 dogs and 225 wolves).
193. Joy Halverson & Christopher J. Basten, A PCR Multiplex and Database for Forensic DNA Identification of Dogs, 50 J. Forensic Sci. 352 (2005); Marilyn A. Menotti-Raymond et al., An STR Forensic Typing System for Genetic Individualization of Domestic Cat (Felis catus) Samples, 50 J. Forensic Sci. 1061 (2005); L.H.P. van de Goor et al., Population Studies of 16 Bovine STR Loci for Forensic
basic reproductive patterns of the species in question. The simple product rule (Section IV) assumes that the sexually reproducing species mates at random with regard to the STR loci used for typing. When that is the case, the usual STR alleles and loci can be regarded as independent. But if mating is nonrandom, as occurs when individuals within a species are selectively bred to obtain some property such as coat color, body type, or behavioral repertoire, or as occurs when a species exists in geographically distinct subpopulations, the inheritance of loci may no longer be independent. Because it cannot be assumed a priori that a crime scene sample originates from a mixed-breed animal, inbreeding normally must be accounted for.194
A different approach is called for if the species is not sexually reproducing. For example, many plants, some simple animals, and bacteria reproduce asexually. With asexual reproduction, most offspring are genetically identical to the parent. All the individuals that originate from a common parent constitute, collectively, a clone. The major source of genetic variation in asexually reproducing species is mutation. When a mutation occurs, a new clonal lineage is created. Individuals in the original clonal lineage continue to propagate, and two clonal lineages now exist where before there was one. Thus, in species that reproduce asexually, genetic testing distinguishes clones, not individuals; hence, the product rule cannot be applied to estimate genotype frequencies for individuals. Rather, the frequency of a particular clone in a population of clones must be determined by direct observation. For example, if a rose thorn found on a suspect’s clothing were to be identified as originating from a particular cultivar of rose, the relevant question becomes how common that variety of rose bush is and where it is located in the community.
In short, the approach for estimating a genotype frequency depends on the reproductive pattern and population genetics of the species. In cases involving unusual organisms, a court will need to rely on experts with sufficient knowledge of the species to verify that the method for estimating genotype frequencies is appropriate.
Purposes, 125 Int’l J.L. Med. 111 (2009). But see People v. Sutherland, 860 N.E.2d 178 (Ill. 2006) (conflicting expert testimony on the representativeness of dog databases); Barbara van Asch & Filipe Pereira, State-of-the-Art and Future Prospects of Canine STR-Based Genotyping, 3 Open Forensic Sci. J. 45 (2010). (recommending collaborative efforts for standardization and additional development of population databases)..
194. This can be done either by using the affinal model for a structured population or by using the probability of a match to a littermate or other closely related animal in lieu of the general random-match probability. See Sutherland, 860 N.E.2d 178 (describing such testimony).
adenine (A). One of the four bases, or nucleotides, that make up the DNA molecule. Adenine binds only to thymine. See nucleotide.
affinal method. A method for computing the single-locus profile probabilities for a theoretical subpopulation by adjusting the single-locus profile probability, calculated with the product rule from the mixed population database, by the amount of heterogeneity across subpopulations. The model is appropriate even if there is no database available for a particular subpopulation, and the formula always gives more conservative probabilities than the product rule applied to the same database.
allele. In classical genetics, an allele is one of several alternative forms of a gene. A biallelic gene has two variants; others have more. Alleles are inherited separately from each parent, and for a given gene, an individual may have two different alleles (heterozygosity) or the same allele (homozygosity). In DNA analysis, the term is applied to any DNA region (even if it is not a gene) used for analysis.
allelic ladder. A mixture of all the common alleles at a given locus. Periodically producing electropherograms of the allelic ladder aids in designating the alleles detected in an unknown sample. The positions of the peaks for the unknown can be compared to the positions in a ladder electropherogram produced near the time when the unknown was analyzed. Peaks that do not match up with the ladder require further analysis.
Alu sequences. A family of short interspersed elements (SINEs) distributed throughout the genomes of primates.
amplification. Increasing the number of copies of a DNA region, usually by PCR.
amplified fragment length polymorphism (AMP-FLP). A DNA identification technique that uses PCR-amplified DNA fragments of varying lengths. The DS180 locus is a VNTR whose alleles can be detected with this technique.
antibody. A protein (immunoglobulin) molecule, produced by the immune system, that recognizes a particular foreign antigen and binds to it; if the antigen is on the surface of a cell, this binding leads to cell aggregation and subsequent destruction.
antigen. A molecule (typically found in the surface of a cell) whose shape triggers the production of antibodies that will bind to the antigen.
autoradiograph (autoradiogram, autorad). In RFLP analysis, the X-ray film (or print) showing the positions of radioactively marked fragments (bands) of DNA, indicating how far these fragments have migrated, and hence their molecular weights.
autosome. A chromosome other than the X and Y sex chromosomes.
band. See autoradiograph.
band shift. Movement of DNA fragments in one lane of a gel at a different rate than fragments of an identical length in another lane, resulting in the same pattern “shifted” up or down relative to the comparison lane. Band shift does not necessarily occur at the same rate in all portions of the gel.
base pair (bp). Two complementary nucleotides bonded together at the matching bases (A and T or C and G) along the double helix “backbone” of the DNA molecule. The length of a DNA fragment often is measured in numbers of base pairs (1 kilobase (kb) = 1000 bp); base-pair numbers also are used to describe the location of an allele on the DNA strand.
Bayes’ theorem. A formula that relates certain conditional probabilities. It can be used to describe the impact of new data on the probability that a hypothesis is true. See the chapter on statistics in this manual.
bin, fixed. In VNTR profiling, a bin is a range of base pairs (DNA fragment lengths). When a database is divided into fixed bins, the proportion of bands within each bin is determined and the relevant proportions are used in estimating the profile frequency.
binning. Grouping VNTR alleles into sets of similar sizes because the alleles’ lengths are too similar to differentiate.
bins, floating. In VNTR profiling, a bin is a range of base pairs (DNA fragment lengths). In a floating bin method of estimating a profile frequency, the bin is centered on the base-pair length of the allele in question, and the width of the bin can be defined by the laboratory’s matching rule (e.g., ±5% of band size).
blind proficiency test. See proficiency test.
capillary electrophoresis. A method for separating DNA fragments (including STRs) according to their lengths. A long, narrow tube is filled with an entangled polymer or comparable sieving medium, and an electric field is applied to pull DNA fragments placed at one end of the tube through the medium. The procedure is faster and uses smaller samples than gel electrophoresis, and it can be automated.
ceiling principle. A procedure for setting a minimum DNA profile frequency proposed in 1992 by a committee of the National Academy of Sciences. One hundred persons from each of 15 to 20 genetically homogeneous populations spanning the range of racial groups in the United States are sampled. For each allele, the higher frequency among the groups sampled (or 5%, whichever is larger) is used in calculating the profile frequency. Compare interim ceiling principle.
chip. A miniaturized system for genetic analysis. One such chip mimics capillary electrophoresis and related manipulations. DNA fragments, pulled by
small voltages, move through tiny channels etched into a small block of glass, silicon, quartz, or plastic. This system should be useful in analyzing STRs. Another technique mimics reverse dot blots by placing a large array of oligonucleotide probes on a solid surface. Such hybridization arrays are useful in identifying SNPs and in sequencing mitochondrial DNA.
chromosome. A rodlike structure composed of DNA, RNA, and proteins. Most normal human cells contain 46 chromosomes, 22 autosomes and a sex chromosome (X) inherited from the mother, and another 22 autosomes and one sex chromosome (either X or Y) inherited from the father. The genes are located along the chromosomes. See also homologous chromosomes.
coding and noncoding DNA. The sequence in which the building blocks (amino acids) of a protein are arranged corresponds to the sequence of base pairs within a gene. (A sequence of three base pairs specifies a particular one of the 20 possible amino acids in the protein. The mapping of a set of three nucleotide bases to a particular amino acid is the genetic code. The cell makes the protein through intermediate steps involving coding RNA transcripts.) About 1.5% of the human genome codes for the amino acid sequences. Another 23.5% of the genome is classified as genetic sequence but does not encode proteins. This portion of the noncoding DNA is involved in regulating the activity of genes. It includes promoters, enhancers, and repressors. Other gene-related DNA consists of introns (that interrupt the coding sequences, called exons, in genes and that are edited out of the RNA transcript for the protein), pseudogenes (evolutionary remnants of once-functional genes), and gene fragments. The remaining, extragenic DNA (about 75% of the genome) also is noncoding.
CODIS (combined DNA index system). A collection of databases on STR and other loci of convicted felons, maintained by the FBI.
complementary sequence. The sequence of nucleotides on one strand of DNA that corresponds to the sequence on the other strand. For example, if one sequence is CTGAA, the complementary bases are GACTT.
control region. See D-loop.
cytoplasm. A jelly-like material (80% water) that fills the cell.
cytosine (C). One of the four bases, or nucleotides, that make up the DNA double helix. Cytosine binds only to guanine. See nucleotide.
database. A collection of DNA profiles.
degradation. The breaking down of DNA by chemical or physical means.
denature, denaturation. The process of splitting, as by heating, two complementary strands of the DNA double helix into single strands in preparation for hybridization with biological probes.
deoxyribonucleic acid (DNA). The molecule that contains genetic information. DNA is composed of nucleotide building blocks, each containing a base (A, C, G, or T), a phosphate, and a sugar. These nucleotides are linked together in a double helix—two strands of DNA molecules paired up at complementary bases (A with T, C with G). See adenine, cytosine, guanine, thymine.
diploid number. See haploid number.
D-loop. A portion of the mitochrondrial genome known as the “control region” or “displacement loop” instrumental in the regulation and initiation of mtDNA gene products. Two short “hypervariable” regions within the D-loop do not appear to be functional and are the sequences used in identity or kinship testing.
DNA polymerase. The enzyme that catalyzes the synthesis of double-stranded DNA.
DNA probe. See probe.
DNA profile. The alleles at each locus. For example, a VNTR profile is the pattern of band lengths on an autorad. A multilocus profile represents the combined results of multiple probes. See genotype.
DNA sequence. The ordered list of base pairs in a duplex DNA molecule or of bases in a single strand.
DQ. The antigen that is the product of the DQA gene. See DQA, human leukocyte antigen.
DQA. The gene that codes for a particular class of human leukocyte antigen (HLA). This gene has been sequenced completely and can be used for forensic typing. See human leukocyte antigen.
EDTA. A preservative added to blood samples.
electropherogram. The PCR products separated by capillary electrophoresis can be labeled with a dye that glows at a given wavelength in response to light shined on it. As the tagged fragments pass the light source, an electronic camera records the intensity of the fluorescence. Plotting the intensity as a function of time produces a series of peaks, with the shorter fragments producing peaks sooner. The intensity is measured in relative fluorescent units and is proportional to the number of glowing fragments passing by the detector. The graph of the intensity over time is an electropherogram.
electrophoresis. See capillary electrophoresis, gel electrophoresis.
endonuclease. An enzyme that cleaves the phosphodiester bond within a nucleotide chain.
environmental insult. Exposure of DNA to external agents such as heat, moisture, and ultraviolet radiation, or chemical or bacterial agents. Such exposure
can interfere with the enzymes used in the testing process or otherwise make DNA difficult to analyze.
enzyme. A protein that catalyzes (speeds up or slows down) a reaction.
epigenetic. Heritable changes in phenotype (appearance) or gene expression caused by mechanisms other than changes in the underlying DNA sequence. Epigenetic marks are molecules attached to DNA that can determine whether genes are active and used by the cell.
ethidium bromide. A molecule that can intercalate into DNA double helices when the helix is under torsional stress. Used to identify the presence of DNA in a sample by its fluorescence under ultraviolet light.
exon. See coding and noncoding DNA.
fallacy of the transposed conditional. See transposition fallacy.
false match. Two samples of DNA that have different profiles could be declared to match if, instead of measuring the distinct DNA in each sample, there is an error in handling or preparing samples such that the DNA from a single sample is analyzed twice. The resulting match, which does not reflect the true profiles of the DNA from each sample, is a false match. Some people use “false match” more broadly, to include cases in which the true profiles of each sample are the same, but the samples come from different individuals. Compare true match. See also match, random match.
gel, agarose. A semisolid medium used to separate molecules by electrophoresis.
gel electrophoresis. In RFLP analysis, the process of sorting DNA fragments by size by applying an electric current to a gel. The different-size fragments move at different rates through the gel.
gene. A set of nucleotide base pairs on a chromosome that contains the “instructions” for controlling some cellular function such as making an enzyme. The gene is the fundamental unit of heredity; each simple gene “codes” for a specific biological characteristic.
gene frequency. The relative frequency (proportion) of an allele in a population.
genetic drift. Random fluctuation in a population’s allele frequencies from generation to generation.
genetics. The study of the patterns, processes, and mechanisms of inheritance of biological characteristics.
genome. The complete genetic makeup of an organism, including roughly 23,000 genes and many other DNA sequences in humans. Over three billion nucleotide base pairs comprise the haploid human genome.
genotype. The particular forms (alleles) of a set of genes possessed by an organism (as distinguished from phenotype, which refers to how the genotype expresses itself, as in physical appearance). In DNA analysis, the term is
applied to the variations within all DNA regions (whether or not they constitute genes) that are analyzed.
genotype, multilocus. The alleles that an organism possesses at several sites in its genome.
genotype, single locus. The alleles that an organism possesses at a particular site in its genome.
guanine (G). One of the four bases, or nucleotides, that make up the DNA double helix. Guanine binds only to cytosine. See nucleotide.
haploid number. Human sex cells (egg and sperm) contain 23 chromosomes each. This is the haploid number. When a sperm cell fertilizes an egg cell, the number of chromosomes doubles to 46. This is the diploid number.
haplotype. A specific combination of linked alleles at several loci.
Hardy-Weinberg equilibrium. A condition in which the allele frequencies within a large, random, intrabreeding population are unrelated to patterns of mating. In this condition, the occurrence of alleles from each parent will be independent and have a joint frequency estimated by the product rule. See independence, linkage disequilibrium.
heteroplasmy, heteroplasty. The condition in which some copies of mitochondrial DNA in the same individual have different base pairs at certain points.
heterozygous. Having a different allele at a given locus on each of a pair of homologous chromosomes. See allele. Compare homozygous.
homologous chromosomes. The 44 autosomes (nonsex chromosomes) in the normal human genome are in homologous pairs (one from each parent) that share an identical set of genes, but may have different alleles at the same loci.
homozygous. Having the same allele at a given locus on each of a pair of homologous chromosomes. See allele. Compare heterozygous.
human leukocyte antigen (HLA). Antigen (foreign body that stimulates an immune system response) located on the surface of most cells (excluding red blood cells and sperm cells). HLAs differ among individuals and are associated closely with transplant rejection. See DQA.
hybridization. Pairing up of complementary strands of DNA from different sources at the matching base-pair sites. For example, a primer with the sequence AGGTCT would bond with the complementary sequence TCCAGA on a DNA fragment.
independence. Two events are said to be independent if one is neither more nor less likely to occur when the other does.
interim ceiling principle. A procedure proposed in 1992 by a committee of the National Academy of Sciences for setting a minimum DNA profile frequency. For each allele, the highest frequency (adjusted upward for sampling
error) found in any major racial group (or 10%, whichever is higher), is used in product-rule calculations. Compare ceiling principle.
intron. See coding and noncoding DNA.
kilobase (kb). A measure of DNA length (1000 bases).
likelihood ratio. A measure of the support that an observation provides for one hypothesis as opposed to an alternative hypothesis. The likelihood ratio is computed by dividing the conditional probability of the observation given that one hypothesis is true by the conditional probability of the observation given the alternative hypothesis. For example, the likelihood ratio for the hypothesis that two DNA samples with the same STR profile originated from the same individual (as opposed to originating from two unrelated individuals) is the reciprocal of the random-match probability. Legal scholars have introduced the likelihood ratio as a measure of the probative value of evidence. Evidence that is 100 times more probable to be observed when one hypothesis is true as opposed to another has more probative value than evidence that is only twice as probable.
linkage. The inheritance together of two or more genes on the same chromosome.
linkage equilibrium. A condition in which the occurrence of alleles at different loci is independent.
locus. A location in the genome, that is, a position on a chromosome where a gene or other structure begins.
mass spectroscopy. The separation of elements or molecules according to their molecular weight. In the version being developed for DNA analysis, small quantities of PCR-amplified fragments are irradiated with a laser to form gaseous ions that traverse a fixed distance. Heavier ions have longer times of flight, and the process is known as matrix-assisted laser desorption-ionization time-of-flight mass spectroscopy. MALDI-TOF-MS, as it is abbreviated, may be useful in analyzing STRs.
match. The presence of the same allele or alleles in two samples. Two DNA profiles are declared to match when they are indistinguishable in genetic type. For loci with discrete alleles, two samples match when they display the same set of alleles. For RFLP testing of VNTRs, two samples match when the pattern of the bands is similar and the positions of the corresponding bands at each locus fall within a preset distance. See match window, false match, true match.
match window. If two RFLP bands lie within a preset distance, called the match window, that reflects normal measurement error, they can be declared to match.
microsatellite. Another term for an STR.
minisatellite. Another term for a VNTR.
mitochondria. A structure (organelle) within nucleated (eukaryotic) cells that is the site of the energy-producing reactions within the cell. Mitochondria contain their own DNA (often abbreviated as mtDNA), which is inherited only from mother to child.
molecular weight. The weight in grams of 1 mole (approximately 6.02 × 1023 molecules) of a pure, molecular substance.
monomorphic. A gene or DNA characteristic that is almost always found in only one form in a population. Compare polymorphism.
multilocus probe. A probe that marks multiple sites (loci). RFLP analysis using a multilocus probe will yield an autorad showing a striped pattern of 30 or more bands. Such probes are no longer used in forensic applications.
multilocus profile. See profile.
multiplexing. Typing several loci simultaneously.
mutation. The process that produces a gene or chromosome set differing from the type already in the population; the gene or chromosome set that results from such a process.
nanogram (ng). A billionth of a gram.
nucleic acid. RNA or DNA.
nucleotide. A unit of DNA consisting of a base (A, C, G, or T) and attached to a phosphate and a sugar group; the basic building block of nucleic acids. See deoxyribonucleic acid.
nucleus. The membrane-covered portion of a eukaryotic cell containing most of the DNA and found within the cytoplasm.
oligonucleotide. A synthetic polymer made up of fewer than 100 nucleotides; used as a primer or a probe in PCR. See primer.
paternity index. A number (technically, a likelihood ratio) that indicates the support that the paternity test results lend to the hypothesis that the alleged father is the biological father as opposed to the hypothesis that another man selected at random is the biological father. Assuming that the observed phenotypes correctly represent the phenotypes of the mother, child, and alleged father tested, the number can be computed as the ratio of the probability of the phenotypes under the first hypothesis to the probability under the second hypothesis. Large values indicate substantial support for the hypothesis of paternity; values near zero indicate substantial support for the hypothesis that someone other than the alleged father is the biological father; and values near unity indicate that the results do not help in determining which hypothesis is correct.
pH. A measure of the acidity of a solution.
phenotype. A trait, such as eye color or blood group, resulting from a genotype. point mutation. See SNP.
polymarker. A commercially marketed set of PCR-based tests for protein polymorphisms.
polymerase chain reaction (PCR). A process that mimics DNA’s own replication processes to make up to millions of copies of short strands of genetic material in a few hours.
polymorphism. The presence of several forms of a gene or DNA characteristic in a population.
population genetics. The study of the genetic composition of groups of individuals.
population structure. When a population is divided into subgroups that do not mix freely, that population is said to have structure. Significant structure can lead to allele frequencies being different in the subpopulations.
primer. An oligonucleotide that attaches to one end of a DNA fragment and provides a point for more complementary nucleotides to attach and replicate the DNA strand. See oligonucleotide.
probe. In forensics, a short segment of DNA used to detect certain alleles. The probe hybridizes, or matches up, to a specific complementary sequence. Probes allow visualization of the hybridized DNA, either by a radioactive tag (usually used for RFLP analysis) or a biochemical tag (usually used for PCR-based analyses).
product rule. When alleles occur independently at each locus (Hardy-Weinberg equilibrium) and across loci (linkage equilibrium), the proportion of the population with a given genotype is the product of the proportion of each allele at each locus, times factors of two for heterozygous loci.
proficiency test. A test administered at a laboratory to evaluate its performance. In a blind proficiency study, the laboratory personnel do not know that they are being tested.
prosecutor’s fallacy. See transposition fallacy.
protein. A class of biologically important molecules made up of a linear string of building blocks called amino acids. The order in which these components are arranged is encoded in the DNA sequence of the gene that expresses the protein. See coding DNA.
pseudogenes. Genes that have been so disabled by mutations that they can no longer produce proteins. Some pseudogenes can still produce noncoding RNA.
quality assurance. A program conducted by a laboratory to ensure accuracy and reliability.
quality audit. A systematic and independent examination and evaluation of a laboratory’s operations.
quality control. Activities used to monitor the ability of DNA typing to meet specified criteria.
random match. A match in the DNA profiles of two samples of DNA, where one is drawn at random from the population. See also random-match probability.
random-match probability. The chance of a random match. As it is usually used in court, the random-match probability refers to the probability of a true match when the DNA being compared to the evidence DNA comes from a person drawn at random from the population. This random true match probability reveals the probability of a true match when the samples of DNA come from different, unrelated people.
random mating. The members of a population are said to mate randomly with respect to particular genes of DNA characteristics when the choice of mates is independent of the alleles.
recombination. In general, any process in a diploid or partially diploid cell that generates new gene or chromosomal combinations not found in that cell or in its progenitors.
reference population. The population to which the perpetrator of a crime is thought to belong.
relative fluorescent unit (RFU). See electropherogram.
replication. The synthesis of new DNA from existing DNA. See polymerase chain reaction.
restriction enzyme. Protein that cuts double-stranded DNA at specific base-pair sequences (different enzymes recognize different sequences). See restriction site.
restriction fragment length polymorphism (RFLP). Variation among people in the length of a segment of DNA cut at two restriction sites.
restriction fragment length polymorphism (RFLP) analysis. Analysis of individual variations in the lengths of DNA fragments produced by digesting sample DNA with a restriction enzyme.
restriction site. A sequence marking the location at which a restriction enzyme cuts DNA into fragments. See restriction enzyme.
reverse dot blot. A detection method used to identify SNPs in which DNA probes are affixed to a membrane, and amplified DNA is passed over the probes to see if it contains the complementary sequence.
ribonucleic acid (RNA). A single-stranded molecule “transcribed” from DNA. “Coding” RNA acts as a template for building proteins according the sequences in the coding DNA from which it is transcribed. Other RNA transcripts can be a sensor for detecting signals that affect gene expression, a switch for turning genes off or on, or they may be functionless.
sequence-specific oligonucleotide (SSO) probe. Also, allele-specific oligonucleotide (ASO) probe. Oligonucleotide probes used in a PCR-associated detection technique to identify the presence or absence of certain base-pair sequences identifying different alleles. The probes are visualized by an array of dots rather than by the electrophoretograms associated with STR analysis.
sequencing. Determining the order of base pairs in a segment of DNA.
short tandem repeat (STR). See variable number tandem repeat.
single-locus probe. A probe that only marks a specific site (locus). RFLP analysis using a single-locus probe will yield an autorad showing one band if the individual is homozygous, two bands if heterozygous. Likewise, the probe will produce one or two peaks in an STR electrophoretogram.
SNP (single nucleotide polymorphism). A substitution, insertion, or deletion of a single base pair at a given point in the genome.
SNP chip. See chip.
Southern blotting. Named for its inventor, a technique by which processed DNA fragments, separated by gel electrophoresis, are transferred onto a nylon membrane in preparation for the application of biological probes.
thymine (T). One of the four bases, or nucleotides, that make up the DNA double helix. Thymine binds only to adenine. See nucleotide.
transposition fallacy. Also called the prosecutor’s fallacy, the transposition fallacy confuses the conditional probability of A given B [P(A|B)] with that of B given A [P(B|A)]. Few people think that the probability that a person speaks Spanish (A) given that he or she is a citizen of Chile (B) equals the probability that a person is a citizen of Chile (B) given that he or she speaks Spanish (A). Yet, many court opinions, newspaper articles, and even some expert witnesses speak of the probability of a matching DNA genotype (A) given that someone other than the defendant is the source of the crime scene DNA (B) as if it were the probability of someone else being the source (B) given the matching profile (A). Transposing conditional probabilities correctly requires Bayes’ theorem.
true match. Two samples of DNA that have the same profile should match when tested. If there is no error in the labeling, handling, and analysis of the samples and in the reporting of the results, a match is a true match. A true match establishes that the two samples of DNA have the same profile. Unless the profile is unique, however, a true match does not conclusively prove that the two samples came from the same source. Some people use “true match” more narrowly, to mean only those matches among samples from the same source. Compare false match. See also match, random match.
variable number tandem repeat (VNTR). A class of RFLPs resulting from multiple copies of virtually identical base-pair sequences, arranged in succession at a specific locus on a chromosome. The number of repeats varies from
individual to individual, thus providing a basis for individual recognition. VNTRs are longer than STRs.
window. See match window.
X chromosome. See chromosome.
Y chromosome. See chromosome.
Forensic DNA Interpretation (John Buckleton et al. eds., 2005).
John M. Butler, Fundamentals of Forensic DNA Typing (2010).
Ian W. Evett & Bruce S. Weir, Interpreting DNA Evidence: Statistical Genetics for Forensic Scientists (1998).
William Goodwin et al., An Introduction to Forensic Genetics (2d ed. 2011).
David H. Kaye, The Double Helix and the Law of Evidence (2010).
National Research Council Committee on DNA Forensic Science: An Update, The Evaluation of Forensic DNA Evidence (1996).
National Research Council Committee on DNA Technology in Forensic Science, DNA Technology in Forensic Science (1992).
The President’s DNA Initiative, Forensic DNA Resources for Specific Audiences, available at www.dna.gov/audiences/.


















































































