
4
Comparative Regulatory and Legal Frameworks
Stem cell therapies are regulated differently in various countries around the world, with some countries offering stem cell therapies that are not available elsewhere. In some cases, offering a particular stem cell therapy can have a positive economic impact for the country in which it is offered because patients are willing to travel in hopes of finding effective therapeutic options. However, a lack of regulations concerning stem cell treatments or a lack of enforcement of such regulations could cause patients harm and damage the industry. For a summary of issues related to these concerns, see Box 4-1.
- In the United States, stem cell therapies are regulated as biologics and are subject to premarket approval under the risk-based approach to approving cellular and tissue-based products. Treatments that are “minimally manipulated” are exceptions to this regulation, but this phrase could use further clarification. (Riley)
- European Union law states that cells or tissues that are “substantially manipulated” are subject to regulation; however, an exemption is given to member states in the non-routine case of custom-made treatments for individual patients. (Bianco)
- The Japanese government has made innovation, including innovation in stem cell research, a national priority, and new laws regarding regenerative medicine products are expected to provide a more efficient path to the translation of these therapies to the clinic. (Miyata)
- Mexico has benefited economically from offering stem cell therapies that are not available in other countries; however, regulations for treating patients with cells and tissues that are based on evidence could help to address challenges relating to therapy, safety, and efficacy. (Arellano)
- While stem cell therapies offered in China must be registered, there are not yet approved quality control guidelines available. (Zhou)
REGULATORY AND LEGAL FRAMEWORKS IN THE UNITED STATES
If regulations for stem cell therapies were only just now being developed in the United States, they would look much different than the ones that exist today, said Margaret Foster Riley, professor of law at the University of Virginia School of Law. The FDA uses regulations to ensure the safety and effectiveness of drugs, biological products, and medical devices under the Biologics Control Act of 1902 and the Food, Drug, and Cosmetic Act of 1938. These statutes have been amended and modernized over time, but they are still a “remarkably ill fit” for stem cell technology, Riley said.
In 1997, the FDA issued a risk-based, tiered approach to regulating cellular and tissue-based products (FDA, 1997). As a result, human cells, with the exception of those deemed minimally manipulated and used for autologous treatments, would be subject to FDA premarket approval as biologics. These products are and the stem cell clinics that offer them, are subject to registration and good practice requirements, Riley said. Thus far, the FDA has not recognized many products as falling into the minimally manipulated category, despite earlier expectations that more and more products would do so over time.
A tissue reference group at the FDA is used to determine the applicability of premarket approval requirements, Riley said. The tissue reference group provides “a single reference point for product specific questions received by FDA concerning jurisdiction and applicable regulation of human cells, tissues and cellular and tissue-based products” (FDA, 2013). However, because of confidential information and other considerations that limit access to the full data, it can be difficult to fully understand the reasoning behind the tissue reference group’s recommendations, Riley said.
Experimental Treatments
The expansion of access to and expedited approval of investigational treatments being evaluated in clinical trials in the United States resulted largely from the acquired immunodeficiency syndrome (AIDS) activism movement, Riley explained (Young, 1988). To receive such access, patients need to have a serious or immediately life-threatening disease or condition, with no comparable or satisfactory alternative therapy available. The potential benefit to the patient must outweigh the risks, and providing the investigational drug for the requested use must not interfere with the initiation, conduct, or completion of clinical investigations, Riley said.
Regulating stem cell therapies is a challenge. The FDA has sent warning letters to several clinics that claim to offer treatments, prevention, or even cures using stem cell therapies that are not approved. For example, the FDA took action against Regenerative Sciences, LLC, which manufactured a product called Regenexx™, which consisted of autologous mesenchymal stem cells that were manipulated outside of the body and injected back into patients with orthopedic injuries (FDA, 2009).1
The Federal Trade Commission
A core task of the Bureau of Consumer Protection of the Federal Trade Commission (FTC) is to protect U.S. consumers from false, misleading, or deceptive advertising, said David Vladeck, a former director of the bureau. The agency focuses on advertising that promotes inaccurate health claims, including dietary supplements and food products that are said to provide health benefits, as well as various kinds of cures for diseases and conditions such as cancer, diabetes, alcoholism, and obesity. The FTC works closely with the FDA in pursuing this mission. Under the Food, Drug, and Cosmetic Act, advertising for treatments based on stem cells that are not approved by the FDA is a serious infraction of the law. The FDA has the option of proceeding against an entity touting stem
__________________
1In February 2014 the U.S. Circuit Court of Appeals for the District of Columbia ruled that the cell mixture used for this procedure contained both a drug and biologic, that it was more than minimally manipulated, and that it qualified as interstate commerce and thus, it was subject to the regulation and approval of the FDA. See United States v. Regenerative Sciences, LLC, No. 12-5254 F.3d (D.C. Cir. 2014).
cell usage for unapproved uses either through civil or criminal law enforcement. Depending on the circumstances, civil actions can begin with warning letters. If the warnings are not heeded, actions can escalate to lawsuits, seizures, and other orders directly regulating the conduct of those involved.
In cases that the FDA judges to pose more of a threat to human health, the agency also may proceed criminally, and it has done that with respect to unproven uses of stem cells, Vladeck said. In September 2012 two individuals who ran several businesses advertising and promoting the use of stem cells, dietary supplements, and vaccines to treat amyotrophic lateral sclerosis, MS, and Parkinson’s disease pled guilty to their charges.2 “The point of this prosecution for the FDA was to send a warning [to] those who would follow in their footsteps and make it clear that recurrences of this would be met by further criminal enforcement actions by the FDA,” Vladeck said. In 2013 the FDA obtained a guilty plea from an assistant professor of pathology and laboratory medicine at the Medical University of South Carolina for “causing the introduction of stem cells into interstate commerce without the approval of the FDA.”3 The laboratory for which the defendant worked was authorized to conduct research on kidney cancer, but it was not permitted to use, harvest, or process stem cells for other purposes, and the defendant sold stem cells to unauthorized recipients, Vladeck said.
Generally, but not always, the FTC has principal responsibility with respect to advertising, Vladeck said, while both the FTC and the FDA have authority over Internet marketing. With emergent scientific issues like stem cell therapies, the FTC will look to the FDA for guidance on scientific issues. However, the FTC enforces its own statute, not the Food, Drug, and Cosmetic Act. With respect to health claims, the FTC can act only if it can show that a claim is false or deceptive; the issue is thus whether there is adequate scientific substantiation to support the claim.
For the FDA, the issue of advertising comes down to a yes-or-no question, Vladeck explained: Has the FDA approved the therapy? If not,
__________________
2FDA, September 7, 2012: Convictions entered in two separate Texas cases involving stem cells. See http://www.fda.gov/ICECI/CriminalInvestigations/ucm319377.htm (accessed March 9, 2014).
3FDA, July 31, 2013: South Carolina man enters plea to introducing stem cells into interstate commerce. See http://www.fda.gov/ICECI/CriminalInvestigations/ucm363400.htm (accessed March 9, 2014).
it is an unapproved use, and the advertised sale of the product is illegal. By contrast, for the FTC the issue is whether a claim is scientifically valid. If it is, then the FTC cannot act, but if it is not, then the FTC has a role to play. These can be difficult cases for the FTC, Vladeck admitted. The FTC will be less inclined to pursue situations in which there are respectable arguments to be made on both sides of the issue. But if the science is inadequate, risks are high, and little evidence suggests benefits, the FTC would be more interested in intervening.
Enforcement
To date, the FTC has not brought up charges in an enforcement case regarding the advertising of unapproved or unsubstantiated stem cell therapies. The issue has not become prominent at the agency, Vladeck said, adding that the FTC received “no or virtually no complaints” about stem cell advertising during his tenure at the agency. It may be that only a limited number of U.S. patients are using clinics offering these therapies in other countries, that patients may be unaware of the complaint mechanisms that are available or think that complaining would be futile, or that U.S. patients may not believe they had been misled or harmed, Vladeck said.
A typical website has a litany of disclaimers tailored to escape U.S. regulation, Vladeck said. Examples of disclaimers include
- “The statements, treatments, and other information on this website have not been evaluated by the U.S. FDA,”
- “The stem cell protocols we offer are not approved in the United States as treatments, therapies, drugs, new drugs, or investigational drugs,”
- “We do not claim that our treatment protocols are approved by the U.S. FDA or proven to be effective in the United States for any condition that appears on this site or for any other condition,”
- “There could be significant and unknown risks associated with adult stem cell treatment as long-term studies have not yet been performed,” and
- “Very few randomized controlled trials of adult stem cells have been performed; therefore, no guarantee of safety or effectiveness is made or implied.”
None of these disclaimers would necessarily defeat an FTC claim, Vladeck said, because the sites claim that these therapies have some scientific grounding and hold out some promise of effectiveness. “But these are certainly sophisticated operators who understand the dynamics of U.S. law,” he added.
Whether a company is physically in the United States does not matter to the FTC, Vladeck said. If a company is selling a product in the United States, the FTC can pursue it. But offshore activities can raise problems for the FTC. If activities are illegal in both the United States and in the host state, it is easier for the FTC to pursue a company because it has counterpart agencies in most countries with which it can work. Where it is legal in the host country, the FTC cannot depend on the cooperation of the host country. That does not mean, however, that the FTC is powerless; it can still bring charges against advertising in the United States even if the advertising takes place over the Internet. The media outlets that convey such information can be put on notice that the FTC believes the information is false or misleading, and a media outlet that disseminates false and misleading information may have liability under U.S. laws.
The FTC bases enforcement actions on several sources of information. It receives about two million original complaints from consumers each year, gets referrals from other agencies, including the FDA, and receives complaints from trusted sources such as academics and experts in the field. However, stem cells have not been the source of widespread complaints.
There should be a balance between paternalism and individual choice, Vladeck said. “At the FTC, our job is not to prevent people from making bad choices. It is to ensure that people do not make bad choices on the basis of bad information.” Government needs to play a greater role in making sure that understandable information flows to patients who can make their own decisions. Websites are being constructed by “very skilled marketers who are selling hope,” Vladeck said. “We have all seen the mythology and the lure of miracle cures.” One year acai berries or green tea may be heralded as promising treatments. Currently it is stem cells that are getting such attention.
REGULATORY AND LEGAL FRAMEWORKS IN ITALY
Most of the regulatory framework that dictates how to use stem cells in Italy derives from a set of European regulations, based largely on the
FDA guiding principles, that have the force of law, said Paolo Bianco, professor and director of anatomic pathology and chief of the Stem Cell Laboratory in the Department of Molecular Medicine at the Sapienza University of Rome and the Umberto I University Hospital. A 2007 regulation, called 1394/2007, covers advanced therapy medicinal products (EMA, 2007). It states that cells that are either cultured or differentiated in culture or used in a location and for a function that is not the same as the same cells in the donor’s body and these cells are referred to as advanced therapy medicinal products. This regulation sets the use of stem cells apart from the use of any other kind of directly transplanted cells or tissues.
Cells that are engineered are defined in the 1394/2007 regulation as cells that have been “substantially manipulated” or modified in such a way that “biological characteristics, physiological functions or structural properties relevant for the intended regeneration, repair or replacement are achieved.” The use of extensive culture, the use of chemicals to induce any kind of differentiation, or the direction to a tissue organ other than the one from which they were derived is considered by European regulators as making cells “substantially manipulated” and thus subject to regulation, Bianco said. The regulation also clearly states what is not considered substantial manipulation, including actions such as centrifugation, the addition of antibiotics or antimicrobials, and irradiation (EMA, 2007).
The regulation also includes a hospital exemption which allows the use of stem cell treatments under certain conditions, Bianco said. The regulation states,
Advanced therapy medicinal products which are prepared on a non-routine basis according to specific quality standards, and used within the same Member State in a hospital under the exclusive professional responsibility of a medical practitioner, in order to comply with an individual medical prescription for a custom-made product for an individual patient, should be excluded from the scope of this Regulation whilst at the same time ensuring that relevant Community rules related to quality and safety are not undermined. (EMA, 2007)
In April 2013 the Senate in Italy voted to reclassify the infusion of mesenchymal stem cells as a transplants procedure, which abrogated the
need for good manufacturing process (GMP) procedures, Bianco said. This took cell therapies out of the jurisdiction of the Italian Medicines Agency made it easier for patients with a variety of unrelated diseases to be treated, without the requirement of conducting clinical trials or pursuing approval of medicines.
Several factors, such as the hospital exemption and unclear regulation wording that contributes to misunderstandings, have led to gaps in regulation. These gaps have been used in some countries by unauthorized stem cells clinics, Bianco said. For example, the private Stamina Foundation in Italy gained substantial support from the public for stem cell therapies they promoted to treat a wide range of medical conditions (Abbott, 2013). The Foundation advertised cures for diseases ranging from autism and psoriasis to urinary incontinence, but with a specific focus on severe lethal neurodegenerative diseases, Bianco said. The therapeutic procedure involved the intravenous or intrathecal injection of mesenchymal stem cells that were said to have been differentiated into nerve cells, though how these were isolated and cultured was largely unknown until 2013 when the methodology was released to an expert review committee (Abbott, 2014).
In May 2013, after support from the national and international scientific community, the Italian lower house countered the April 2013 vote of the Senate, Bianco said, and GMP procedures and vigilance by the Italian Medicines Agency were restored as mandatory. Nearly $4 million was allocated for a government-supervised clinical trial to evaluate the Stamina Foundation stem cell therapy. However, following the review of the procedures by an expert committee, in October 2013 the health minister halted the trial (Abbott, 2014). It was reported that the expert committee had concerns over pathogen screening, the method for generating mesenchymal stem cells, and the use of inappropriate assays to identify the cells, among other things (Abbott, 2014).
Remaining Concerns
The problems with stem cell regulation have not been eradicated, Bianco said. Other interests were exerting pressure on the Italian government at the same time. For example, during the Stamina controversy, the minister received a report claiming that the marketing of mesenchymal stem cells should be allowed with no trial of efficacy but only after a small Phase I trial indicating, but not demonstrating, safety had been performed.
Of 361 clinical trials using intravenous infusion of mesenchymal stem cells for autism and urinary incontinence, only two have been completed and reported on ClinicalTrials.gov as Phase III trials, Bianco said, and both of these had negative results. Indeed, the only product based on mesenchymal cells that has ever been approved is one for which two Phase III trials were completed with negative results. Additionally, many clinical trials are not reported to ClinicalTrials.gov or other registries, making it difficult to determine the incidence of adverse events in all trials, Bianco said.
The problem is not simply a scientific or medical problem, Bianco said. The translation of science to the marketplace needs to be considered, not just the translation of science to medicine.
REGULATORY AND LEGAL FRAMEWORKS IN JAPAN
Japan has a very similar regulatory framework to that of the United States and the European Union, said Toshio Miyata, executive director of the Health and Global Policy Institute in Japan. Prime Minister Shinzo Abe has been eager to push forward the development of Japanese innovation, including stem cell research, especially since the Nobel Prize–winning work of Shinya Yamanaka (Nobel Media, 2013; Takahashi et al., 2007). The prime minister has submitted a bill to establish a new independent administrative agency that would ensure integrated management of research, Miyata said.
As of December 2012, Japan had approved only two regenerative medicine products, compared with 20 in Europe, 14 in South Korea, 9 in the United States, and 6 in other regions, including Southeast Asia and Australia, Miyata said. Sixty-five products were undergoing clinical research under guidelines established by the Ministry of Health, Labor, and Welfare (MHLW). Under the Pharmaceutical Affairs Law, which provides regulation for drugs and devices in Japan, the Pharmaceuticals and Medical Devices Agency (PMDA) conducts conformity audits and scientific reviews and reports to the MHLW, which issues approvals. Essentially, the PMDA acts as the technical arm of the MHLW, which has the ultimate responsibility for policies and administrative measures.
In 2012 the PMDA established the Office of Cellular and Tissue-Based Products under the Center for Product Evaluation, Miyata said. Under a new 5-year clinical trial activation plan, the number of pharmaceutical trials has been increasing in Japan. Clinical trials in Japan go
through a series of phases that are similar to those in the United States and the European Union.
As part of its Life Innovation Project for Health Care, Japan has been developing a strategy for drugs and medical devices aimed at achieving practical application of projects originating in Japan, Miyata said. The country also is creating early phase clinical trial centers for innovative drugs and medical devices. In addition, it is developing guidance based on regulatory science to streamline review for innovative drugs, medical devices, and biologics. Among these innovative products are platelets and retinal cells derived from induced pluripotent stem cells (Kamao et al., 2014; Nakamura et al., 2014).
The Japanese government has been considering the unique characteristics of regenerative medicine products in establishing regulations. For example, it has labeled therapies that use stem cells as “regenerative medicine products” and has introduced a new definition of regenerative and cellular therapeutic products that sets them apart from pharmaceuticals and medical devices. It also has developed an approval system for earlier commercialization of regenerative medicine products, with the introduction of a tentative approval after which safety and efficacy are further confirmed. Pharmaceutical regulation is a high hurdle for academia, Miyata said, and the reform of the regulations will create a unique category for regenerative medicines.4
The new approval system for the commercialization of cellular therapy products would move approval from the end of clinical trials to a stage intermediate between the confirmation of probable benefit and safety and marketing with further confirmation of efficacy and safety to follow (see Figure 4-1). The post-market phase would include informed consent and safety measures such as period reports and record retention, Miyata said.
Japan has experienced various controversies surrounding stem cell treatments. In 2010 a Korean patient traveled to Japan to receive a stem cell therapy not offered in Korea and then died of a pulmonary embolism (Cyranoski, 2010). In response, a bill for ensuring the safety of the
__________________
4In November 2013 the National Diet (Japan’s legislative branch) passed legislation to revise the Pharmaceutical Affairs Law and approve the new Regenerative Medicine Law with the goal of bringing safe stem cell therapies to patients more quickly. See Japan’s bold initiative in regenerative medicine and who the big winners might be, http://www.marketwatch.com/story/japans-bold-initiative-in-regenerative-medicine-and-who-the-bigwinners-might-be-2014-03-03 (accessed March 9, 2014).
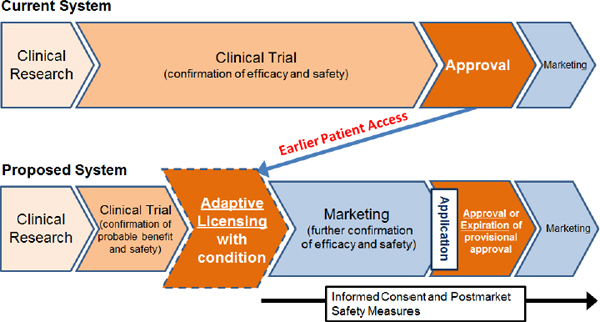
FIGURE 4-1 A new approval system under consideration in Japan would create earlier patient access to cellular therapy products.
NOTE: Probable benefit—Confirmation of efficacy with a small patient population; Safety—Earlier detection and evaluation of adverse events.
SOURCE: Toshio Miyata, IOM, NAS, and ISSCR workshop presentation on November 18, 2013.
procedures has been proposed. It outlines a number of safety measures including prior notification to authorities to ensure the safety of the treatment, establishing a permitting and notification system for cell-processing facilities, and obtaining informed consent, and it also calls for the protection of personal information and the reporting of adverse events to authorities, Miyata said.
REGULATORY AND LEGAL FRAMEWORKS IN MEXICO
The medical tourism industry is an important business in Mexico, which has been a popular destination for foreign patients who are seeking stem cell treatments, said María de Jesús Medina Arellano, an attorney from Universidad Autonoma de Nayarit who has studied health law and human rights in Mexico. The main consumers of services have been Americans and Canadians who travel to Mexico to obtain treatments that are either unavailable in or are less expensive than in their home countries.
Many private companies in Mexico offer stem cell treatments. However, with just three exceptions, none of the treatments offered in Mexico have been evaluated in Federal Commission for the Protection Against Sanitary Risk (in Spanish, Comisión Federal para la Protección contra Riesgos Sanitarios, or COFEPRIS) registered clinical trials. Yet, many of these treatments are offered as proven and effective therapies. COFEPRIS is an independent government authority of the Ministry of Health that has the exclusive statutory authority to oversee the inspection, approval, and authorization of activities concerning the use, storage, and transplantation of umbilical cord blood or derived human stem cells. However, COFEPRIS does not have standards or guidelines to implement and enforce when evaluating, authorizing, and monitoring research and therapeutic activities involving human tissues and cells, Arellano said (Arellano, 2012).
Mexico has a constitutional right to access to health care, Arellano noted. Based on that right, the General Health Law governs biomedical research and, arguably, is applicable to the clinical use of experimental stem cell treatments. In addition, the Biomedical Research Regulation gives the conditions required to perform clinical trials on human subjects, including research involving the use of human organs, tissues, and derivatives. However, none of these regulations specifically applies to stem cell therapies. The General Health Law prohibits false advertising of treatments or clinical interventions that are not supported by scientific evidence and at least five official Mexican standards prohibit and regulate the advertising of medical products and services, Arellano said. However, the regulations have little effective oversight, and COFEPRIS reportedly lacks financial as well as well-trained human resources.
Changes in the Laws
In December 2012 the law governing stem cell therapies changed, following lobbying from private medical enterprises in Mexico, Arellano said. Previously the law had prohibited any kind of commercialization of organs, tissues, and cells. The law now states that the procurement, extraction, analysis, derivation, preservation, transportation, and supply of organs, tissues, and stem cells are not considered acts of commerce.
This change in the law has created problems in Mexico, Arellano reported. An article in the June 2012 issue of Medical Tourism Magazine states that “Mexico offers an alternative treatment destination” for stem cell therapies that have not been approved in a prospective recipients
home country (George, 2012). But without proof of safety and efficacy, Arellano said, it will be difficult to protect patients undertaking what is essentially experimental medicine.
Regulations should be flexible, Arellano acknowledged, but there should also be evidence that treatments are going to work. To date, clinics have not been asked or required to share information about their offerings. Doing so would provide an opportunity for the government to effectively oversee stem cell science applications while fostering innovation in the area, Arellano said. Both the government and the medical sector need greater transparency in order to build confidence among patients. International regulation or harmonization in the area of stem cells, despite skepticism about international regulations, could help address many of the challenges Mexico faces.
REGULATORY AND LEGAL FRAMEWORKS IN CHINA
The Ministry of Science and Technology, the Chinese Academy of Sciences (CAS), and the National Science Foundation of China all support stem cell research in China, with funding that totaled a little less than $500 million over a 5-year period, said Qi Zhou, director of the State Key Laboratory of Reproductive Biology and vice director of the Institute of Zoology at CAS. For example, the Innovation 2020 Stem Cell Research Project of CAS involves about 3,000 scientists, including staff scientists and postdoctoral fellows, working on stem cell research at almost 30 different institutions. Altogether, said Zhou, about 200 hospitals and many institutions are working on stem cell research and cell therapy, and China now publishes the second largest number of stem cell publications, behind researchers in the United States.
New regulations have recently been put into place in China, Zhou said, with the Ministry of Science and Technology and the Ministry of Health publishing ethical guidelines and regulations for human embryonic stem cell research. This has been in response to the increasing use of unapproved stem cell treatments in clinics and skepticism over their therapeutic efficacy (Cyranoski, 2009, 2012; Dobkin et al., 2006). These new guidelines and standards have been disseminated in Chinese and described in several published papers.
In October 2011, a National Stem Cell Research Supervision and Coordination Committee was formed to coordinate all stem cell research and technology. The director of the committee is the Minister of Science
and Technology, and the National Science Foundation, the Ministry of Health, the Ministry of Education, and other ministries are represented on the committee. A committee also exists for industrial applications of stem cells and regenerative medicine.
Oversight of Stem Cell Treatments
Since June 2012, all organizations conducting stem cell therapy have had to register their treatments, Zhou said. Guidelines on quality control and preclinical research on stem cell preparations have been published but have not yet been approved by the government. Organizations involved in these guidelines include the National Health and Family Planning Commission of China, the China Food and Drug Administration, the National Leading Group of Clinical Stem Cells Study, and the National Stem Cell Experts Committee. The guidelines cover the isolation, purification, culture, amplification, modification, differentiation, cryopreservation and resuscitation, and in vivo implantation of stem cells, including embryonic stem cells, induced pluripotent stem cells, mesenchymal stem cells, hematopoietic stem cells, and other progenitor cells or precursor cells. Security evaluations include the detection of bacteria and fungi mycoplasma, the detection of endogenous and exogenous viral agents, and reagent detection, while validity evaluations include cell identification tests, examining the pluripotency of stem cells, the detection of stem cell–specific markers, cell activity assays, and animal models.
Clinical trials can be halted at each level, Zhou said. The Chinese government has been proactive in this regard, notifying and halting illegal stem cell therapies, as they are identified. This occurred, for instance, with illegal stem cell therapies that were being provided by a hospital in Jilin province. However, physicians in China generally have more freedom to pursue treatments than do physicians in the United States, Zhou said, because there are fewer enforcement mechanisms in China.
“China would like to cooperate with the world to promote stem cell applications,” Zhou concluded.
SUGGESTED IMPROVEMENTS FOR REGULATION
The questions being addressed by regulatory science will be important to the future development of stem cell therapies. This idea has been criticized because of the perception that regulation will thwart in-
novation, but regulatory science can develop pathways that allow innovation in the best safety framework, Riley said.
Patient safety is key. More transparency is needed for understanding clinical trials so that patients understand the treatments and the experimental design, Arellano said. The most important goal for stem cell therapy, Bianco said, is to make sure that unproven therapies are not marketed to patients. The FTC’s role is to avert harm, and a way must be found to get clear information to consumers, Vladeck said.
If unproven therapies are being offered to consumers in the United States, federal regulatory agencies need to know about it, Vladeck emphasized. The FTC needs to understand the magnitude of the problem, including the risks to patients in both physical and financial terms. The FTC would respond well to comments or overtures by scientific groups, professional bodies, or patients that are pointing to a serious problem, he said, but the FTC needs information about patient experiences. For example, how many patients are traveling outside of the United States for these therapies, and what are the treatment outcomes? Are patients satisfied with the results?
Regulators need to work more closely with scientists, even if they are involved in commercial enterprises, Bianco said. Regulatory agencies need to work more rapidly and efficiently on the processing and approval of everything they regulate. Interactive discussions with the FDA convened by organizations the like California Institute for Regenerative Medicine (CIRM) are important ways for bringing academia and industry together to discuss stem cell therapy regulations, said Ellen G. Feigal of CIRM. A good next step, Arellano said, would be to invite policy makers to these kinds of discussions in order to get them involved in the science and regulation of new therapies along with more people from developing countries who are working on these treatments.
International harmonization for stem cell therapies can be difficult in a domain characterized by complexity and powerful commercial interests, Riley said, but harmonization could be pursued through international societies working together with government agencies. Multi-site international clinical trials would be one avenue toward greater harmonization, she said. Miyata supported the simultaneous global development of Phase I trials in Japan, the United States, the European Union, and other countries, along with the promotion of global clinical trials.
Accelerating regulatory science initiatives, with early communication and coordination among regulatory bodies in different countries, harmonizing guidelines and regulations, and the adoption of simultane-
ous approval in different countries would also be helpful, Miyata said. Perhaps working through groups like the United Nations Educational Scientific and Cultural Organization is also a possibility, Arellano suggested. Trying to harmonize on the basis of ethical issues from a global perspective will be difficult, Bianco said, but there is a role for the NAS and similar institutions around the world.
















