
Summary
Patients and health care providers expect quality-assured, safe, and effective medicines that are properly labeled for the purposes for which they have been approved. A key objective of medicines regulatory authorities is to promote and protect public health by ensuring the quality, safety, and efficacy of medicines and by ensuring that they are properly labeled based on robust science. Fulfilling this mission in a time of rapid scientific change, increasing complexity of medicines, and globalization of production and product supply chains has presented regulatory authorities with multiple challenges while also opening doors to opportunities in the form of greater regulatory cooperation and information sharing among the regulators.
It was in this context that the U.S. Food and Drug Administration’s (FDA’s) Office of International Programs (now the Office of Global Policy and Strategy) asked the National Academies of Sciences, Engineering, and Medicine to convene an ad hoc committee of experts to conduct a landscape review of the tools (e.g., recognition and other regulatory arrangements) being used by regulators to help them oversee the quality, safety, and efficacy of medicines throughout the product lifecycle (from initial development through marketing approval and distribution). Along with this landscape, the committee’s Statement of Task outlined requests to analyze interagency recognition and other forms of reliance in terms of major challenges and opportunities as well as potential risks and benefits. To carry out its charge, the committee held a series of information-gathering sessions along with two open meetings at which the committee heard from representatives of industry, patient groups, international organizations, and regulatory authorities. These informants’ comments were supplemented by written feedback from
a targeted group of regulators; a comprehensive literature search; and online information, mainly in the form of government-issued reports. With this collective input, as well as recognition of the common public health mission of all regulatory authorities, the committee synthesized two key messages. These key messages serve as the lens through which the committee’s conclusion and recommendations should be viewed:
- Regulation through recognition and reliance arrangements is now a 21st-century best regulatory practice. Accordingly, regulators, regardless of human, technical, and financial resources, need to make increased use of reliance and cooperation with other trusted regulators, as no regulator has the resources it needs to meet all of its public health responsibilities.
- Impediments to regulators entering into and using such informal and formal recognition and reliance arrangements to help them obtain the information they need for their regulatory decision making should be removed.
In addition to and based on its one conclusion, the committee formulated six recommendations, the first of which includes a strategy for improving regulatory cooperation1 that places the public’s health at the center of collaboration efforts. The strategy targets multiple stakeholder groups with an expressed interest in public health and patient care. Within this strategy, each group is called on to play a role in enhancing regulatory cooperation in the form of recognition and reliance arrangements that range from informal, collaborative activities to the most formal—a mutual recognition agreement (MRA). It is safe to say that virtually all regulatory authorities are motivated by a public health mission to safeguard their populations with respect to the medicines coming into, and currently on, the market in their jurisdictions. Additionally, given the rapid movement of people and illnesses across borders in today’s world, it is similarly safe to say that public health is a global concern and that all regulators, regardless of size or financial resources, exercise their responsibilities within that global public health framework as part of a larger global regulatory enterprise. How regulatory authorities carry out their missions and structure their regulatory cooperation activities is often a function of human and financial resources. In this report, and as reflected in the recommended strategy, regulatory authorities are divided into three categories: well-resourced, moderately well-resourced, and lower-resourced.
___________________
1 Regulatory cooperation is considered by the committee to be any formal or informal interaction with another regulator for the purposes of sharing information or working together toward a common goal.
ROLE OF REGULATORY AUTHORITIES
The core functions of regulatory authorities include ensuring that products released for use in their jurisdiction are properly evaluated and meet appropriate standards of quality, safety, and efficacy that are maintained throughout all stages of the product lifecycle and supply chain, including manufacturing, production, packaging, and distribution (WHO, 2019g). These basic functions must be met while enabling timely access to quality-assured products and encouraging innovation (WHO, 2019e). It is industry’s responsibility to comply with rules and scientific and technical standards established for products, and it is the regulator’s responsibility to oversee compliance with these rules and standards (Rönninger et al., 2012) and to work with the larger community to develop new standards as technology and scientific knowledge improve. For medicines (the focus of this report), there are regulations describing compliance activities and good practices across the medicines lifecycle from preclinical research to post-marketing surveillance. Guidance is provided by regulatory authorities at each stage (as shown in Figure S-1), but it is up to industry to interpret the guidance and apply it to their situations. Good laboratory practice (GLP) sets guidelines for ensuring that laboratory data are of high quality and reliable in the preclinical phase; good clinical practice (GCP) guides aspects of studies involving human subjects; good manufacturing practice (GMP) provides guidance for the manufacturing, production, and distribution of medicines; and good pharmacovigilance practice (GPvP) provides guidance for the appropriate oversight of products once they have been released onto the general market. Assuring an overall positive benefit/risk profile to the most appropriate level possible is essential throughout the entire medicine product lifecycle.
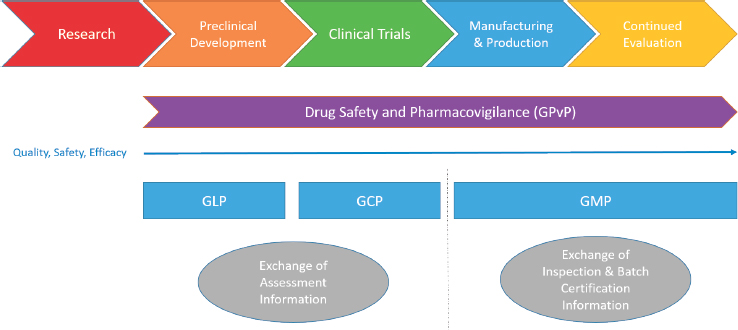
For the purposes of this report, regulatory work products are the administrative documents produced by a regulator as a result of a regulatory authority’s evaluation. These work products might include an inspection report or certificate of compliance for GMP, official batch release, or an assessment report regarding evaluation of the clinical and statistical elements of the clinical data used in the application requesting approval to market a new medicine.
Sponsors of new drugs create a registration dossier (e.g., “marketing authorization application,” “new drug application”) that is submitted to a regulatory authority for assessment regarding possible approval for marketing. It will typically include information from the sponsor’s product development program, such as data purporting to demonstrate the overall positive benefit/risk profile of the product, including its safety and effectiveness in clinical studies; the preclinical laboratory data; and the data surrounding the manufacture of the product. If the product is approved for marketing, regulatory authorities will continue to oversee its manufacture and production and ongoing benefit/risk profile, often taking a risk-based approach to determine the level of intensity of that oversight. Each stage of the product lifecycle presents opportunities for the use of recognition and reliance between and among regulatory authorities. Often, the design and extent of such recognition and reliance are based on the availability of the regulatory authorities’ technical resources.
RECOGNITION AND RELIANCE
As defined by the World Health Organization (WHO), recognition occurs when a regulatory authority accepts the regulatory decision of another authority “as its own decision.” Reliance takes place when a regulatory authority takes into account the work products of another authority (e.g., inspection reports, scientific assessment reports, joint assessment reports produced together with another authority) to help inform the receiving authority’s own regulatory decision, which in the end may differ from the decision made by the initial authority using the same work products. In essence, one could argue that recognition is a subset of reliance, or the “ultimate reliance.” The important point is that even when an authority uses recognition or reliance, it has sovereignty over and responsibility for the regulatory decision it makes for its country—it does not “outsource” its decision making. A regulatory authority’s decision to routinely accept the regulatory decision of another authority or to take into account the work products of another regulator to inform its decision making is the choice and responsibility of that regulatory authority. The decision to do so might be based on a variety of factors, including the context in which a regulatory decision is being made, access to technical or financial resources, and
whether there is sufficient trust and confidence in the decision and/or work product/information of the regulatory authority being relied on.
Different tools to facilitate recognition and reliance across the product lifecycle are available. These are collectively referred to in this report as “arrangements.” Arrangements can be more formal, established and implemented, for example, through Memoranda of Understanding, confidentiality commitments, and MRAs; or they can be informal arrangements through the establishment of ad hoc committees, working parties, and other, more topic-specific working groups (EMA, 2019g). WHO’s draft guidelines for good regulatory practices note that “less formal practices include sharing of information, scientific collaboration, common risk assessments, joint reviews, and development of standards” (WHO, 2016b, p. 27).
Much attention has historically been focused on the use of recognition and reliance arrangements with respect to GMP inspections, while there has been less of a focus on GLP, GCP, and GPvP inspections or other aspects of regulatory decision making. These other areas may represent opportunities for future expansion of arrangements.
UNDERSTANDING THE REGULATORY ENVIRONMENT
In an environment of limited human and financial resources and at a time of unprecedented globalization and societal requests for faster approvals of drugs, medicines regulators have felt pressure to stretch their finite resources. To that end, they have undertaken a wide range of activities geared toward helping each other manage a growing workload by leveraging those limited resources. While a few regulatory authorities have access to higher levels of technical and financial resources, the vast majority of medicines regulatory authorities globally would be considered moderately well- to lower-resourced. In fact, WHO estimates that, based on the criteria in its Global Benchmarking Tool, approximately 100 of the 194 WHO member states do not have a medicines regulatory agency that is technically capacitated to meet even the basic requirements of a medicines regulatory authority (Khadem, 2019). For many regulatory authorities, relying on the work of other trusted regulators is the only way to keep pace with the growing demand for their services. This was the overwhelming message of regulators in Australia, Canada, Singapore, and Switzerland who have joined together through a work-sharing project based on trust and confidence built among the partners. Pilot tests, working groups, and other convening activities designed to build trust and confidence are often essential first steps toward relying on the work of other regulators. If trust and confidence are satisfactorily built, authorities may consider engaging in formal and/or informal recognition or reliance arrangements, the most formal of which are MRAs. Figure S-2 shows how trust and confidence are
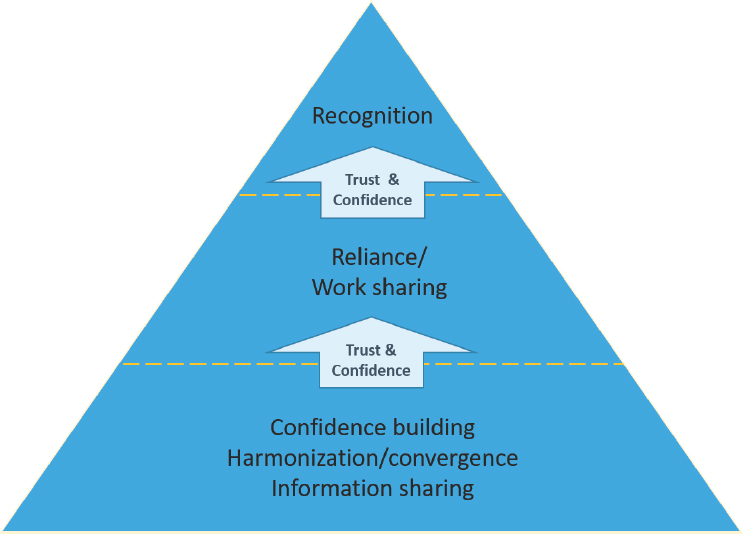
SOURCES: Adapted from a figure created by Dr. Petra Doerr, Swissmedic, and presented by Emer Cooke, World Health Organization (WHO, 2019i).
necessary for all forms of recognition and reliance arrangements (mutual and others).
There are currently 14 MRAs for medicines, listed in Appendix B of this report. While less formal reliance arrangements promote the sharing of information and facilitate fewer redundant inspections and other regulatory activities, MRAs allow regulatory authorities not just to rely on each other’s work products but to recognize them officially as equivalent to their own. However, this is possible only if shared reports are complete and essentially free from redactions. Current medicines regulatory MRAs are limited primarily to GMP reports and official batch release reports, although a few also include GLP reports. To progress beyond GMP and expand the classes of products subject to the GMP MRAs, regulators would likely have to establish high levels of trust and confidence by building a track record of demonstrated equivalence. In the meantime, regulators use or could use other, more agile reliance tools to work with other regulators in managing their growing workloads to help ensure the quality, safety, efficacy, and availability of medicines.
LEVERAGING KEY OPPORTUNITIES TO OVERCOME MEDICINES REGULATORY CHALLENGES
Rapid globalization of drug discovery, development, manufacturing, and delivery has significant implications for public health. The resources required to assure comprehensive oversight of these activities is now an enormous task for even well-resourced national medicines regulatory authorities. All have limited resources to accomplish sufficiently all the tasks they are asked to perform. The ongoing evolution of the science and technologies associated with drug discovery, development, manufacturing, and delivery (e.g., dramatic increases in more complex biological medicines and emerging use of cell and gene therapies) poses additional capacity and expertise challenges for regulatory authorities. These challenges underscore the need for further cooperation and collaboration among authorities; however, achieving that cooperation can be difficult because individual authorities have different histories; legal frameworks; human, technical, and financial resources; and areas of expertise. In confronting the global challenges of medicines regulation, cooperation and collaboration among regulators offer opportunities to share information and increase the transparency of each other’s activities; to share finite resources and address the growing workload resulting from globalization; and to rely on each other’s processes, work products, and decision making via both formal reliance arrangements such as MRAs and other, less formal arrangements.
Based on its extensive deliberations and input from stakeholders representing regulatory authorities, international organizations, industry, and patient groups, the committee agreed that public health must be at the center of all medicines regulatory recognition and reliance arrangements. Additionally, regulatory authorities require the support of industry, patients, and governments to realize the maximum benefits of any recognition or reliance arrangement.
Conclusion: The committee concludes that protecting and promoting public health in a time of globalization and unprecedented advances in technology and medicines—which are mirrored by the growing complexity of medicines and the supply chains for their manufacture and production—is the single greatest challenge facing medicines regulatory authorities today. It is therefore imperative that regulatory authorities at all resource levels—well, moderately well, and lower—find ways to continue or expand on their ability and willingness to work together to maximize the use of their finite resources so they can ensure the quality, safety, efficacy, and availability of medicines for their jurisdictions in both emergency and non-emergency situations.
However, for regulatory authorities to build further on their current recognition and reliance activities, impediments to entering into and using formal and informal recognition and reliance arrangements need to be removed. Some impediments lie within the regulatory authority itself, while others may be external to the agency and influenced by those within policy, industry, or consumer/patient advocacy groups. Each of these stakeholder groups has a role to play in supporting efforts to enhance cooperation among regulatory authorities, with the overarching aim of improving public health. The committee therefore recommends that all regulatory authorities and other key stakeholder groups demonstrate their support for formal and informal medicines regulatory recognition and reliance arrangements using a targeted approach.
Recommendation 1: The committee recommends a strategy that leverages the support of each stakeholder group in the following manner:
- All regulatory authorities, especially those that are well- and moderately well-resourced, should increase information sharing and the transparency of each other’s regulatory activities across the lifecycle of medicines in ways that can facilitate more efficient resource allocation and decision making for all regulators and reduce the burden of redundant regulatory activities on regulators, patients, and industry.
- All regulatory authorities, especially those that are well- and moderately well-resourced, should be allowed to share their work products in essentially unredacted form (i.e., full reports without parts of the report expunged, except for personal privacy information) with other regulatory authorities so assessment and inspection information can be made available to those other regulators, especially those that are lower-resourced, thereby enabling access to quality, usable regulatory information by a greater number of regulatory authorities for addressing global public health needs. In this respect, policy makers, and the U.S. Congress in particular, should weigh the challenges and opportunities involved in empowering their respective medicines regulatory authorities to share complete, unredacted inspection reports (e.g., good manufacturing practice reports) with other regulatory authorities so as to facilitate learning, aid in decision making, reduce the use of limited resources on redundant inspections, decrease the burden on industry of redundant inspections, and strengthen the overall global public heath infrastructure for safe and effective quality medicines.
- Lower-resourced regulatory authorities should consider the risks and benefits of unilateral recognition of the regulatory decisions
-
of trusted regulatory authorities when doing so would facilitate better public health decision making in the context in which the lower-resourced regulatory authority functions.
- Industry should support the recognition and reliance efforts of regulatory authorities by encouraging them to share less redacted or, better, unredacted reports with their trusted regulatory authority partners, and by showing a willingness to share health-related data and/or information relevant to regulatory decision making more publicly to benefit the global public health good and reduce the sharing authority’s own burden of redundant oversight.
- Patient and consumer groups should support the recognition and reliance efforts of medicines regulatory authorities by advocating for a “public health protection and promotion” framing of all such arrangements and for their increased use.
The committee further believes, based on its information gathering and expert opinion, that formal and informal recognition and reliance arrangements are highly effective tools for facilitating cooperation and reliance on the work of other regulatory authorities, when they are established in ways that emphasize public health, maximize efficiencies, reduce the burden of redundancy, and have the potential to benefit both global and national public health by ensuring effective and efficient access to safe and effective quality medicines.
Improving Public Health Through Better Designed MRAs
MRAs are one tool to facilitate the regulatory cooperation that can enable regulators to better fulfill their regulatory public health mandates. MRAs developed through trade agreements—which can include a wide range of commodities such as electrical goods and telecom equipment—can be slow to conclude and costly for administrators, regulators, and negotiators in terms of time and human resources (Correia de Brito et al., 2016). If the primary objective of these agreements that involve medicines is to improve public health, it stands to reason that medicines regulators, with their shared understanding of public health, are key to developing and designing such agreements. MRAs have usually been developed in the context of trade negotiations, with the dual aim of reducing technical barriers to trade and promoting public health. To prioritize what should be the public health aims and focus of these arrangements, the committee believes that medicines regulators, with their public health background, are ideally positioned to increase the scope and substance of interagency reliance. Medicines regulators share a common interest, namely, improving the public’s health through the availability of safe and effective medicines.
That shared, common interest makes medicines regulators well suited to developing, designing, and implementing the substance of such regulatory agreements in the future. Therefore, the committee makes the following recommendation.
Recommendation 2: Policy makers, including lawmakers, should explore empowering regulators to expand the scope and substance of future mutual recognition agreements (MRAs) that address issues related to the safety, efficacy, and manufacturing quality of medicines, and to ensure that these MRAs are designed, developed, and implemented primarily by medicines regulators. Policy makers will also need to ensure that regulators have adequate resources for these tasks.2
Responding to Evolving Science and Technology
MRAs, as currently designed, are not sufficiently agile tools to respond to the rapid pace at which science, technology, and the global medicines regulatory enterprise are all evolving. More agile reliance arrangements would be better suited to meet these challenges. To address the challenges associated with the globalized production of medicines, as well as the growing need to address the medicine requirements during public health emergencies, the committee believes that MRAs and other reliance arrangements should be expanded to include new areas.
Recommendation 3: The committee recommends that regulators consider increasing the current scope of both formal and less formal reliance arrangements, including mutual recognition agreements, and that policy makers encourage regulatory authorities to explore formal and informal opportunities for reliance arrangements with other trusted regulatory authorities that give regulators greater flexibility in responding to challenges that affect their responsibility in overseeing the quality, safety, and efficacy of medicines throughout the medicines’ lifecycle. Potential areas identified for such expansion of scope include good laboratory practice, good clinical practice, and good pharmacovigilance practice inspection reports; preclinical assessment reports;
___________________
2 Murray Lumpkin, Lembit Rago, and Katherine Bond did not fully concur with this recommendation because they believe it still leaves the negotiation, oversight, and finalization of MRAs related to medicines regulation to trade negotiators, rather than empowering medicines regulators to design, develop, conclude, and implement these specific medicines regulatory MRAs on their own. They believe that is the only way to ensure that public health is the sole focus of the negotiation and agreement and that the agreement is negotiated and concluded in the collaborative public health atmosphere that exists among medicines regulators and not the competitive business dynamic that pervades and shades trade negotiations.
bioequivalence assessment reports; and a wider scope of product classes covered by such arrangements.
Better Utilization of the European Union (EU)-US Mutual Recognition Agreement
The EU-US MRA, as currently implemented, narrowly applies only to areas involving GMP and then only to a limited range of products. Some provisions have not been implemented, such as those for inspections conducted outside of the United States and the EU by EU and U.S. authorities, respectively (so-called “third-country” inspections). Those provisions should be implemented immediately. If one regulatory authority can trust the other to perform an inspection within the latter authority’s own jurisdiction, the committee could find no reason to doubt the ability of the latter authority to perform a quality inspection outside its own jurisdiction—especially as these inspections are currently limited to surveillance inspections. The EU-US MRA could be expanded to go beyond its currently limited GMP focus to a broader GMP focus and to other regulatory activities, together with greater product coverage.
Recommendation 4: Regulatory authorities in the United States and the European Union (EU) should immediately implement provisions included in the current EU-US mutual recognition agreement (MRA) (e.g., those regarding so-called “third-country” good manufacturing practice [GMP] inspections). Regulatory authorities also should begin considering the potential for expanding the EU-US MRA to include reliance in areas beyond GMP and a broader scope of products under the current GMP provisions.
Facilitating Information Sharing Among International Medicines Regulators
Without a unified platform and a standard format for reporting, the sharing of assessment and inspection reports can be challenging. The committee recognizes that some regulators currently share assessment, inspection, and other reports with other regulators with whom legally authorized appropriate confidentiality arrangements exist. In instances where such arrangements do not exist, sponsors could explicitly allow regulators to share full assessment reports on specified products with specified regulators. Additionally, a recently concluded confidentiality commitment (CC) between FDA and the EU, known as the Super-CC, includes medicines. The Super-CC provides a mechanism for FDA to share essentially unredacted information with other regulatory authorities under very specific circumstances.
The committee believes that to best meet the public health goals of reliance arrangements, an opportunity exists for FDA and Congress to ensure that redaction practices optimize information sharing. The committee believes that FDA and Congress could reevaluate whether existing confidentiality restrictions are still fit-for-purpose in the 21st-century globalized environment in which medicines products exist and whether modifications are needed to meet the public health goals for these arrangements noted earlier in this report (e.g., protecting and promoting public health; reducing the burden of regulatory redundancy on patients, industry, and regulators; allowing regulators to use the finite human and financial resources they currently have most effectively and efficiently; and helping to bring needed quality medicines to patients domestically and globally as efficiently as possible).
Recommendation 5: Regulatory authorities, with guidance from their governmental leaders, should undertake determining whether current limitations on sharing regulatory work products with other regulatory authorities are still fit-for-purpose to help protect and promote public health; to reduce the burden of regulatory redundancy on patients, industry, and regulators; to allow regulators globally to best utilize the limited technical and financial resources currently available to them to meet their public health mandates; and to bring needed quality medicines to patients domestically and globally as efficiently as possible.
Evaluating Public Health Impacts of Recognition and Reliance Arrangements for Medicines
Evaluating the impacts of formal and informal recognition and reliance arrangements on public health, on the use of regulatory and industry resources, and on the essential regulatory competencies of regulatory authorities is challenging because of a dearth of frameworks, metrics, and data for use in such evaluations. The texts of existing formal and less formal recognition and reliance arrangements generally fail to incorporate review criteria or frameworks, including specific metrics, by which regulatory authorities, governments, and the broader community could evaluate the arrangements’ impacts—most important, their impacts on public health. Creating a results framework with clear indicators/metrics and processes for monitoring and measuring the results of recognition and reliance arrangements could enhance understanding of their various impacts, especially on public health, and enable benefit/risk and cost/benefit analysis of formal and less formal recognition and reliance arrangements over time. Including specific evaluations of public health and other goals, metrics, and mandates in the text of future formal and less formal recognition and reliance
arrangements would contribute to developing a robust body of knowledge regarding their impacts and overall utility.
Recommendation 6: When formal and informal recognition and reliance arrangements are being developed, the regulatory authorities involved should co-create a results framework with clear indicators/metrics and processes for monitoring and measuring the arrangements’ results and impacts to enhance understanding of their public health and other benefits and associated regulatory efficiencies, and enable benefit/risk and cost/benefit analysis of the arrangements over time.
THE WAY FORWARD
As regulators and policy makers contemplate next steps, they would be wise to listen carefully to the views of patients who express their desire for effective and efficient access to quality-assured, safe, and effective medicines and advanced therapeutics that are affordable and equitably available to those who need them. Additionally, regulatory systems could be reviewed with an eye toward effectiveness (i.e., systems that do the right thing) and efficiency (i.e., systems that do the thing right). Given the human, technical, and financial resources currently available to regulatory authorities—even those that are most well-resourced—leveraging the work of other trusted authorities is essential. Eliminating redundant regulatory activities is also essential to having a system that is effective and efficient and able to meet the challenges of the globalized world, in which the products being regulated locally exist today.
At present, recognition and reliance arrangements focus on specific classes of medicines (EMA, 2019f; PIC/S, 2019a). In the future, regulatory authorities might explore the use of more agile recognition and reliance arrangements to share the ideas and perspectives of scientific experts in such areas as advanced therapeutics (e.g., gene therapy) (PIC/S, 2019b). If advanced scientific information could be shared more broadly, regulatory authorities, without local access to such information and perspectives, could also benefit from that information in deciding whether to approve such innovations within their own jurisdictions. Alternatively, regulatory authorities without local access to certain expertise, might explore the degree to which medicines approved by a trusted authority might similarly be recognized for approval in their country or region. In the end, though, regulatory authorities are responsible for regulatory decisions that affect the people in their jurisdiction. The committee believes that having a variety of different recognition and reliance arrangements from which regulators could choose would facilitate the marketing and availability of quality-assured, safe, and effective medicines necessary to safeguard the public’s health.
It is the committee’s view that both formal and informal recognition and reliance arrangements are important regulatory tools for helping regulatory authorities, of all resource levels, address the public health challenges posed by the increasing complexity of medicines and their globalized supply chains. The committee further contends that all medicines regulatory authorities would benefit from increased use of formal and informal recognition and reliance arrangements in conducting activities designed to fulfill their public health mission. It is the committee’s view that regulation through such arrangements can now be considered a 21st-century “best regulatory practice.”














